Du Maladie de Pompe est une maladie de stockage du glycogène d'évolution imprévisible. Les symptômes sont caractérisés par une faiblesse musculaire progressive. En thérapie, des succès grâce à l'administration artificielle de l'enzyme causale ont entre-temps été observés.
Qu'est-ce que la maladie de Pompe?

© designua - stock.adobe.com
Les maladies de stockage sont un groupe hétérogène de maladies dans lesquelles diverses substances sont déposées dans des organes ou des cellules de l'organisme. Les tissus ou organes perdent généralement leur fonction en raison des dépôts. Les substances déposées peuvent être de différents types. Selon la substance, une distinction est faite entre les glycogénoses, les mucopolysaccharidoses, les lipidoses, les sphingolipidoses, les hémosidéroses et les amyloïdoses. Dans la glycogénose, le glycogène est stocké autour des tissus corporels.
Les glucides stockés ne peuvent plus du tout être décomposés ou seulement de manière incomplète ou peuvent être transformés en glucose. La cause est généralement une carence enzymatique des enzymes impliquées dans la conversion du glucose dans le corps. Une maladie du stockage du glycogène est que Maladie de Pompequi aussi Maladie de Pompian ou Déficit acide maltase est appelé. La maladie affecte l'α-glucosidase lysosomale ou la maltase acide sur le gène GAA.
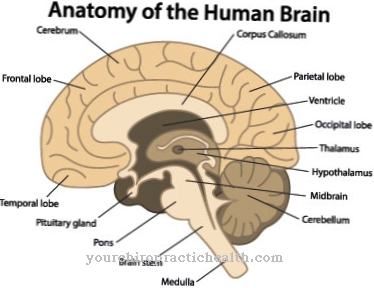
Dans le corps sain, l'enzyme décompose les polysaccharides à longue chaîne des lysosomes en glucose. Chez l'homme, l'enzyme est présente dans tous les tissus. La maladie métabolique est encore principalement perceptible dans les muscles et est donc également incluse dans les myopathies. La maladie rare doit son nom au pathologiste néerlandais Pompe, qui a décrit le phénomène pour la première fois en 1932. En 1954, G.T. Maladie de Cori en tant que maladie de stockage du glycogène de type II.
causes
Dans les années 1960, H.G. Son absence d'α-glucosidase lysosomale comme lien de causalité dans la maladie de Pompe. Cette carence survient sur la base d'un défaut génétique principalement causal qui affecte l'enzyme α-1,4-glucosidase. Cette enzyme est également connue sous le nom de maltase acide et est soit complètement absente, soit a une activité réduite. Cela empêche la dégradation du glycogène sous forme de stockage du sucre dans les muscles. Le glycogène est donc stocké dans les cellules musculaires des lysosomes, où il détruit les cellules musculaires. L'activité restante de l'enzyme est en corrélation avec la gravité de la maladie.
Le type infantile de la maladie de Pompe a une activité inférieure à 1%. Le type juvénile conserve une activité résiduelle allant jusqu'à dix pour cent et le type adulte conserve une activité résiduelle allant jusqu'à 40 pour cent. La maladie est sujette à une transmission autosomique récessive. Les garçons sont touchés aussi souvent que les filles. Le défaut génique causal a été localisé dans la région q25.2-q25.3 du chromosome 17 et est long de 28 kpb.
La maladie est génétiquement hétérogène et a jusqu'à présent été associée à 150 mutations différentes. Les patients sont hétérozygotes composés. La forme infantile est souvent caractérisée par deux mutations sévères. Avec cette forme, il existe un degré élevé de correspondance entre le génotype et l'évolution de la maladie. Ce n'est pas le cas avec la forme adulte. La prévalence est donnée avec des valeurs comprises entre 1: 40 000 et 1: 150 000. Environ 200 personnes ont été diagnostiquées en Allemagne.
Symptômes, maux et signes
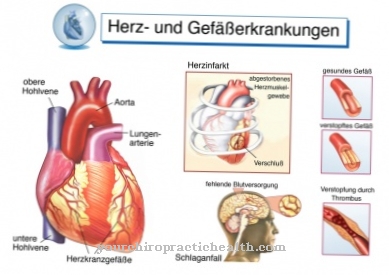
Les principaux symptômes de la maladie de Pompe sont la cardiomégalie et l'insuffisance cardiaque. Il existe également des déficits neurologiques et musculaires. L'épidémie de la maladie de Pompe n'est pas limitée à un certain âge, mais peut toucher toutes les tranches d'âge. La forme infantile survient chez les nourrissons et se termine généralement fatalement au cours de la première année de vie. Dans la plupart des cas, la mort qui survient est la mort par insuffisance cardiaque, qui est due à une cardiomégalie hypertrophique.
Dans la forme infantile, les premiers symptômes apparaissent après deux mois. Les adolescents développent la maladie de Pompe juvénile. Chez l'adulte, la médecine parle de la maladie de Pompe chez l'adulte. Ces formes se manifestent de manière symptomatique par une faiblesse musculaire progressive des muscles respiratoires et des muscles squelettiques du tronc. Le haut des bras peut être touché ainsi que le bassin et les cuisses. Le parcours est imprévisible sous les formes adulte et juvénile. Les parcours difficiles se caractérisent par une perte de respirabilité.
Il y a aussi souvent une perte de mobilité. Des états d'épuisement surviennent. Dans certains cas, des substances se déposent dans les artères et peuvent former un anévrisme dont la rupture peut être fatale. En moyenne, les premiers symptômes deviennent perceptibles peu avant l'âge de 30 ans et correspondent à des difficultés à courir ou à faire de l'exercice. Le diagnostic est généralement posé au milieu des années 30. Environ dix ans plus tard, les personnes touchées sont majoritairement dépendantes d'un fauteuil roulant.
Diagnostic et évolution de la maladie
Le diagnostic de la maladie de Pompe est généralement confirmé par une biopsie musculaire basée sur l'anamnèse. Histologiquement, les dépôts massifs de glycogène dans le muscle peuvent être mis en évidence dans la coloration PAS. Le diagnostic peut tout aussi facilement être ancré dans une mesure de l'activité enzymatique de la maltase acide, comme cela peut être détecté dans les leucocytes avec le test sanguin sec. Les examens génétiques moléculaires peuvent également être utilisés comme moyens de diagnostic. CK, CKMB, LDH, GOT et GPT sont augmentés dans le sang. L'urine est généralement élevée en Glc4.
De nombreux diagnostics différentiels doivent être exclus. Dans la forme infantile, en raison des symptômes prononcés, le diagnostic suspecté peut souvent être assuré par la progression rapide associée à des problèmes respiratoires croissants et au retard du développement moteur. La cardiomégalie peut être confirmée par des résultats radiographiques. Théoriquement, un diagnostic prénatal basé sur un examen du liquide amniotique ou un prélèvement tissulaire est également envisageable.
L'évolution de la maladie de Pompe est généralement plus sévère, plus la maladie se déclare tôt. Néanmoins, la maladie de Pompe est caractérisée par une évolution individuelle et donc en fait imprévisible de la maladie. Des formes légères sont également observées.
Complications
Avec la maladie de Pompe, les personnes touchées souffrent de restrictions et de plaintes importantes dans la vie quotidienne. En premier lieu, il y a des difficultés respiratoires, qui conduisent à une résilience et une fatigue considérablement réduites du patient. De plus, le manque d'oxygène peut également entraîner une perte de conscience, dans laquelle la personne concernée peut éventuellement se blesser à la suite d'une chute.
Les difficultés respiratoires ont également un effet négatif sur le cœur et les autres organes internes, de sorte que des dommages consécutifs irréversibles peuvent survenir aux organes.L'espérance de vie est considérablement réduite et limitée par la maladie de Pompe. Dans le pire des cas, la personne affectée peut mourir de mort cardiaque. Les activités épuisantes ou sportives ne sont toujours pas possibles pour la personne touchée par cette maladie.
Le traitement de la maladie de Pompe est généralement basé sur les symptômes et vise à augmenter l'espérance de vie. Divers médicaments sont utilisés, qui n'entraînent généralement pas de complications particulières. Les symptômes peuvent également être réduits à l'aide de la physiothérapie. S'il existe des limitations psychologiques, un traitement psychologique supplémentaire est nécessaire.
Quand devriez-vous aller chez le médecin?
Toute personne qui souffre de la maladie de Pompe est soit touchée par la maladie héréditaire pendant son enfance, soit présente des symptômes musculaires sévères à l'âge adulte. Les personnes atteintes de glycogénose de type II souffrent d'une perte musculaire croissante. Les muscles respiratoires peuvent également être affectés. Si le défaut génétique n'est pas découvert au début d'un examen de routine, les visites chez le médecin sont à l'ordre du jour à mesure que les symptômes augmentent.
Cependant, le diagnostic peut prendre beaucoup de temps. Premièrement, il existe de nombreuses maladies avec une évolution similaire. Deuxièmement, les tests génétiques ne sont pas la norme en médecine. Troisièmement, la maladie de stockage du glycogène de type II est également une maladie métabolique. De nombreux médecins ne connaissent pas les symptômes de la maladie de Pompe. De plus, il n'y a pas de plainte uniforme pour la maladie de Pompe d'apparition tardive. La maladie est beaucoup plus facile à diagnostiquer chez les enfants.
La plupart des patients adultes consultent un médecin avec une variété de troubles musculaires. Les symptômes décrits peuvent toucher les extrémités, mais aussi les muscles respiratoires ou le cœur. Des organes comme le foie peuvent également être affectés. Le déclin évolue rapidement. En conséquence, au fil du temps, les visites chez le médecin augmentent sans que le diagnostic correct ne soit posé. Une odyssée à travers les pratiques n'est pas rare pour la maladie de Pompe.
Thérapie et traitement
La maladie de Pompe n'est pas encore guérie. Un traitement causal des symptômes n'est pas disponible. Par conséquent, les patients sont principalement traités de manière symptomatique et de soutien. Des formes de thérapie palliative sont recommandées. Cette thérapie comprend des recommandations diététiques et des exercices de respiration ainsi que de la physiothérapie pour renforcer les muscles. Au cours de cela, la ventilation et la nutrition artificielle deviennent nécessaires.
La décision opportune de ces mesures est absolument nécessaire pour prolonger la vie. En termes de nutrition, une alimentation riche en protéines associée à un entraînement d'endurance a fait ses preuves. Depuis 2006, il est possible de fournir artificiellement la protéine recombinante, qui se compose de cellules CHO et est connue sous le nom d'alglucosidase alfa ou myozyme. Le médicament est administré par voie intraveineuse tous les 14 jours. Des succès décisifs ont été observés chez les nourrissons après une administration précoce du médicament, ce qui pourrait assurer la survie.
Il existe des expériences contradictoires avec des enfants plus âgés et il n'existe aucune preuve convaincante de l'efficacité de la forme adulte. Le coût des médicaments peut aller jusqu'à 50 000 euros par an pour un nourrisson et jusqu'à 500 000 euros par an pour un adulte. Un approvisionnement à vie est nécessaire. La réponse des muscles squelettiques à la thérapie est variable. Mais la faiblesse du muscle cardiaque s'améliore. En raison de la barrière hémato-encéphalique, le médicament n'a aucun effet sur les processus pathologiques dans le cerveau.
Les approches thérapeutiques telles que la thérapie génique en sont au stade de l'expérimentation animale. Le transfert de gène a déjà réussi chez la souris. Le traitement avec des chaperons pharmacologiques peut augmenter l'activité résiduelle de la maltase acide, mais jusqu'à présent, il ne peut pas être utilisé de manière pratique. Des soins psychothérapeutiques de soutien pour faire face à la situation sont recommandés pour les familles touchées.
Vous pouvez trouver votre médicament ici
➔ Médicaments contre la douleurPerspectives et prévisions
La maladie de Pompe est une maladie héréditaire incurable qui se traduit généralement par une espérance de vie plus courte. Le pronostic exact dépend de la forme spécifique de la maladie et de l'âge de la personne touchée lorsque la maladie éclate.
Le pronostic est le plus défavorable pour la forme précoce de la maladie de Pompe, qui, si elle n'est pas traitée, entraîne généralement la mort dans les deux ans. Les personnes touchées meurent principalement de pneumonie ou d'insuffisance cardiaque. Le traitement de la maladie par une thérapie enzymatique substitutive améliore considérablement le pronostic. L'espérance de vie des personnes touchées augmente considérablement jusqu'à un âge de plus de dix ans. Cette forme de thérapie étant nouvelle, il n'y a toujours pas de pronostics à long terme.
La forme juvénile de la maladie de Pompe, si elle n'est pas traitée, entraîne généralement la mort avant d'atteindre l'âge adulte. La forme adulte de la maladie de Pompe a le pronostic le plus favorable. Dans tous les cas, l'espérance de vie augmente considérablement avec le traitement. Cependant, les personnes touchées développent généralement certaines limitations telles que des difficultés cognitives et une perte auditive ou même une surdité. De nouvelles options de traitement telles que les thérapies géniques pour la maladie de Pompe sont actuellement en cours de développement et de test, ce qui devrait conduire à un pronostic bien plus favorable.
la prévention
Jusqu'à présent, la maladie de Pompe ne peut être prévenue que par un conseil génétique pendant la phase de planification familiale. Pour les parents d'un enfant atteint, le risque de récidive est de 25%. Après un diagnostic prénatal positif, les futurs parents ont également la possibilité de se faire avorter.
Suivi
En règle générale, les personnes atteintes de la maladie de Pompe ont très peu et très peu de mesures et d'options de soins de suivi disponibles, de sorte qu'elles devraient idéalement consulter un médecin à un stade précoce afin d'éviter la survenue d'autres plaintes et complications. Il ne peut pas guérir indépendamment, de sorte qu'un médecin doit être consulté dès les premiers signes et symptômes de la maladie.
Le processus de récupération repose principalement sur des exercices de physiothérapie ou de physiothérapie. De nombreux exercices peuvent être répétés dans votre propre maison, ce qui favorise considérablement le traitement. L'aide et les soins de sa propre famille sont également très importants, ce qui peut également prévenir la dépression et d'autres troubles psychologiques.
Dans le cas d'un désir existant d'avoir des enfants, les personnes touchées devraient profiter des tests génétiques et des conseils afin d'éviter que la maladie ne se reproduise. Des contrôles et examens réguliers par un médecin sont également nécessaires au cours de la vie. La maladie elle-même ne réduit généralement pas l'espérance de vie du patient. D'autres mesures de suivi ne sont généralement pas disponibles pour la personne concernée.
Tu peux le faire toi-même
Pour les personnes touchées, la maladie de Pompe est un diagnostic très stressant, surtout si la maladie ne se manifeste qu'à un âge avancé. La maladie peut suivre une évolution très individuelle, c'est pourquoi les patients veulent faire le plus possible pour une évolution bénigne de la maladie.
L'essentiel est de prévenir d'autres maladies. Par exemple, les patients doivent s'assurer qu'ils sont bien approvisionnés en oxygène afin de ne pas tomber à cause d'étourdissements ou de subir un accident. De plus, un apport insuffisant en oxygène endommagerait le cœur - et peut-être aussi d'autres organes. Les maladies infectieuses telles que la grippe ou les infections pseudo-grippales doivent également être évitées dans la mesure du possible car elles altèrent également la respiration et affaiblissent gravement le corps et son système immunitaire. Les patients atteints de la maladie de Pompe feraient donc bien de porter une attention particulière à leur système immunitaire. Vous devez vous assurer d'avoir une alimentation fraîche et riche en protéines dans le cadre des recommandations diététiques de votre spécialiste.
L'exercice régulier est également important pour maintenir la force musculaire, en particulier dans les jambes. Surtout, l'entraînement d'endurance a fait ses preuves dans le traitement de la maladie de Pompe. Les kinésithérapeutes traitants apportent l'assistance nécessaire. Le soutien psychologique est utile pour de nombreux patients et leurs proches. Vous pouvez également obtenir de l'aide et des informations auprès de Pompe Deutschland e.V. (www.morbus-pompe.de).






.jpg)



.jpg)

.jpg)