Sous le Syndrome TAR, Anglais Syndrome du rayon sans thrombocytopénie, la médecine comprend un syndrome de malformation, dont les principaux symptômes comprennent la défaillance des rayons et la thrombocytopénie. On pense que la cause du syndrome est une mutation génétique héréditaire. Le traitement au cours des premières années de vie consiste principalement en une transfusion de plaquettes.
Qu'est-ce que le syndrome TAR?

© Reing - stock.adobe.com
le Syndrome TAR est un complexe de multiples malformations qui se manifeste chez les nouveau-nés. Les principaux symptômes du syndrome de malformation héréditaire sont une défaillance bilatérale du rayon et un manque de plaquettes sanguines. En raison des symptômes, le syndrome est parfois daté Syndrome d'aplasie radiale-thrombocytopénie le discours. Le syndrome TAR a jusqu'à présent été décrit dans un peu plus de 100 cas. La prévalence exacte n'est pas connue, mais le complexe de symptômes est considéré comme relativement rare.
Le syndrome a été décrit pour la première fois en 1929. Les Américains H. M. Greenwald et J. Sherman sont considérés comme les premiers à le décrire. Selon la documentation précédente, les femmes sont légèrement plus susceptibles d'être touchées par des malformations que les hommes. En raison de sa rareté, le syndrome n'a pas été entièrement étudié. La recherche sur les causes a donné un succès partiel, mais n'a pas encore été en mesure de fournir une explication suffisante pour l'ensemble du complexe.
causes
En 2007, une cause possible du syndrome TAR a été identifiée qui correspond à une mutation génétique. Les symptômes partiels du complexe sont causés par une microdélétion sur le chromosome 1 dans le locus du gène q21.1. Dans ce contexte, nous parlons du syndrome de délétion 1q21.1. Le chromosome 1 a jusqu'à présent été associé à de nombreuses maladies héréditaires.
Des mutations dans cette localisation génique peuvent déclencher le syndrome d'Usher, la maladie de Gaucher ou la maladie d'Alzheimer, par exemple. Le chromosome 1 est présent sous forme de paire de chromosomes dans toutes les cellules du corps et correspond au plus grand chromosome humain. La mutation associée au syndrome TAR semble nécessairement être présente chez tous les patients. Cependant, la mutation n'explique pas suffisamment les symptômes individuels du syndrome.
Le syndrome TAR est considéré comme une maladie héréditaire. Une accumulation familiale a été observée dans les cas documentés jusqu'à présent. L'hérédité semble être un héritage autosomique récessif et avoir lieu avec une variabilité relativement grande dans la manifestation.
Symptômes, maux et signes
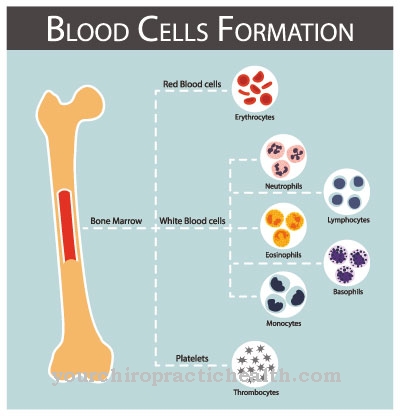
Tous les patients atteints du syndrome TAR ont une thrombocytopénie. Le manque de plaquettes conduit à une tendance accrue à saigner. Ils diminuent particulièrement au cours des deux premières années de vie. Au cours des premiers mois, des saignements intracrâniens peuvent survenir, ce qui peut entraîner un retard moteur ou mental. Bilatéralement, tous les patients atteints du syndrome manquent également de rayons.
Le pouce de la personne affectée est là, mais il ne fonctionne que de manière anormale. Il y a souvent une déviation radiale de la main, qui se manifeste par une déformation de la main du club. Le cubitus de tous les patients TAR est raccourci et partiellement plié. Environ un tiers des patients sont dépourvus d'humérus, qui est généralement également raccourci et a un effet dysplasique. Les articulations du coude, de l'épaule et de la main sont limitées dans leur mobilité.
Dans certains cas, il y a également des changements dans le sang. Dans les deux tiers des cas, les leucocytes sont fortement augmentés. Il existe souvent une allergie ou une intolérance au lait de vache qui favorise la diarrhée ou aggrave la thrombocytopénie. Chez environ la moitié de tous les patients, les symptômes sont associés à une dysplasie des membres inférieurs.
En particulier, la dysplasie de la hanche, la coxa valga, la subluxation de l'articulation du genou ou la dysplasie rotulienne avec luxation sont des symptômes courants. Le genou peut être raidi. Les positions des pieds et des orteils sont souvent anormales. Beaucoup de personnes atteintes souffrent également d'une petite taille ou d'une malformation cardiaque au sens d'une tétralogie de Fallot ou d'une communication interauriculaire. L'œil a souvent un ptosis ou un glaucome.
Diagnostic et évolution de la maladie
Dans les premiers mois de la vie, le médecin observera une tendance aux saignements et à la thrombopénie chez les patients TAR, qu'il doit différencier de l'anémie de Fanconi dans un diagnostic différentiel. En imagerie aux rayons X, le syndrome TAR est particulièrement évident dans le non-alignement des rayons des deux côtés et les désalignements qui en résultent.
En termes de diagnostic différentiel, le syndrome de Holt-Oram et le syndrome de Roberts doivent également être considérés. Dès que les deux premières années de vie se sont écoulées, le pronostic des patients atteints du syndrome TAR est plutôt favorable. Dans des cas individuels, le pronostic est basé sur des symptômes d'accompagnement tels que la malformation cardiaque.
Complications
Diverses malformations se produisent dans le syndrome TAR. D'abord et avant tout, les malformations entraînent une tendance à saigner significativement accrue. Les personnes touchées souffrent de saignements sévères, même avec des blessures très légères et mineures, qui ne peuvent être arrêtées facilement. Les saignements surviennent souvent au niveau des gencives ou du nez et ont un effet très négatif sur la qualité de vie de la personne concernée.
De plus, un retard mental peut survenir en raison du syndrome TAR. Les patients dépendent très souvent de l'aide d'autres personnes dans leur vie et ne peuvent pas faire beaucoup de choses quotidiennes par eux-mêmes. La mobilité des épaules et des mains est également considérablement limitée par le syndrome, car l'humérus est absent. De plus, cela peut entraîner une malformation cardiaque ou une gêne pour les yeux.
Le syndrome est généralement associé à une espérance de vie réduite. Les parents ou proches souffrent également souvent de troubles psychologiques ou de dépression. Le traitement symptomatique du syndrome TAR n'entraîne généralement pas de complications. Malheureusement, toutes les plaintes ne peuvent pas être complètement limitées.
Quand devriez-vous aller chez le médecin?
Dans la plupart des cas, avec le syndrome TAR, la personne touchée aura besoin d'une évaluation médicale et d'un traitement. Il ne peut y avoir de guérison indépendante, de sorte que la personne atteinte de cette maladie dépend toujours d'un diagnostic médical. Plus tôt le syndrome est reconnu, meilleure sera l'évolution de la maladie. Puisqu'il s'agit d'une maladie héréditaire, aucune guérison complète ne peut avoir lieu. Si la personne touchée par le syndrome souhaite avoir des enfants, un conseil génétique peut également être utilisé.
En cas de syndrome TAR, un médecin doit être consulté si la personne concernée souffre d'un retard mental sévère. En règle générale, les patients dépendent de l'aide des autres dans leur vie. La mobilité de la personne touchée peut également être limitée par le syndrome TAR, de sorte qu'une visite chez un médecin est nécessaire. Il n'est pas rare que les organes internes soient affectés par divers défauts. Le diagnostic du syndrome TAR peut être posé par un médecin généraliste ou un pédiatre. Pour un traitement ultérieur, une visite chez un spécialiste est nécessaire.
Thérapie et traitement
Le syndrome TAR ne peut être traité ni de manière causale ni spécifique. Jusqu'à présent, seuls les traitements symptomatiques sont disponibles. Les malformations ne peuvent pas être corrigées dans les premières années de la vie en raison de la tendance à saigner. Dans les derniers stades de la vie, les interventions chirurgicales reconstructives peuvent corriger les rayons manquants et les multiples malpositions. Il est essentiel de prévenir tout saignement ou hémorragie au cours des premières années de vie.
Le but de la thérapie initiale est avant tout de réduire les conséquences importantes de la maladie. Les thrombopénies sévères au cours des premières années de vie nécessitent des transfusions de plaquettes. En soi, il n'y a pas de trouble du développement moteur. Les limitations neurologiques sont également rares. Le développement mental est discret. Tous les retards sont donc au mieux la conséquence d'un saignement intracrânien, qui doit être évité par les transfusions.
Lorsque la thrombocytopénie s'est calmée, un traitement chirurgical plastique est effectué. Ces mesures sont accompagnées de [[[étapes de traitement physiothérapeutique]], qui visent à assurer un développement moteur parfait. À l'âge adulte, les patients ne dépendent souvent plus d'aucune mesure de traitement et mènent une vie largement normale avec une qualité de vie illimitée.
Vous pouvez trouver votre médicament ici
➔ Médicaments pour le traitement des plaies et des blessuresla prévention
Les causes définitives du syndrome TAR n'ont pas encore été élucidées. Pour cette raison, le syndrome ne peut pas encore être évité. Cependant, tout indique que les facteurs génétiques jouent un rôle dans le syndrome. Par conséquent, le conseil génétique pour les personnes touchées peut être largement décrit comme une mesure préventive.
Suivi
Les soins de suivi du syndrome TAR dépendent du type et de la gravité des malformations. Après une intervention chirurgicale, qui est une option pour les malformations mineures, le patient a besoin de soins de suivi approfondis. En cas de saignement aigu, des soins immédiats à la clinique sont nécessaires. Le patient a alors besoin de quelques jours de repos.
Un examen de suivi final vise à déterminer les autres mesures de traitement. Les personnes atteintes du syndrome TAR doivent consulter régulièrement leur médecin pour clarifier leur état de santé actuel. Les complications médicales ne sont pas toujours apparentes pour le patient, en particulier en cas d'hémorragie interne. En cas de thrombopénie sévère, une transfusion sanguine peut être nécessaire, réalisée en clinique et généralement associée à un débriefing.
Le patient à son tour a besoin de repos et de protection. L'hospitalisation est généralement indiquée. Les patients souffrant du syndrome TAR ont besoin d'un médecin spécialiste. Le médecin responsable est généralement l'interniste ou le médecin généraliste déjà impliqué dans le traitement. L'hospitalisation à long terme est logique pour les plaintes chroniques. Le patient doit également contacter un physiothérapeute et d'autres spécialistes. Un soutien psychologique pour le patient peut également être nécessaire.
Tu peux le faire toi-même
Le syndrome TAR ne peut être traité que de manière symptomatique. Le patient doit faire attention aux signes avant-coureurs et informer le médecin afin qu'une transfusion sanguine puisse être effectuée rapidement. Après une telle transfusion, le corps est affaibli et il est important d'assurer une alimentation équilibrée qui aide le corps à produire du sang.
Après le traitement chirurgical des malformations, le repos et l'alitement s'appliquent. Le patient doit prendre soin des plaies selon les instructions du médecin pour éviter l'inflammation et d'autres complications. En cas de malformations des membres, un traitement physiothérapeutique peut également être nécessaire. Les personnes touchées peuvent faire de la physiothérapie à domicile et améliorer la coordination des membres affectés grâce à un exercice régulier.
Si ces mesures n'atteignent pas le résultat souhaité, le médecin doit être consulté. Le syndrome TAR étant une affection extrêmement rare, un médecin spécialiste doit reprendre le traitement. Il est conseillé de rechercher d'autres personnes dans les forums Internet, car il n'y a que quelques groupes d'entraide pour la maladie. Enfin, il est important de faire les visites nécessaires chez le médecin afin d'éviter des complications graves. Le syndrome TAR doit être étroitement surveillé en raison de toute transfusion sanguine.


.jpg)