Au Déficit en α-N-acétylgalactosaminidase c'est une maladie du stockage lysosomal qui survient très rarement. Il est divisé en une forme juvénile et une forme adulte.
Qu'est-ce que le déficit en a-N-acétylgalactosaminidase?

Le déficit en a-N-acétylgalactosaminidase ou Déficit en alpha-N-acétylgalactosaminidase s'appelle aussi en médecine Maladie de Schindler, Maladie de Schindler ou Maladie de Kanzaki désigné. Il s'agit d'une maladie de stockage lysosomal extrêmement rare, dont l'hérédité est autosomique récessive. Les médecins font la différence entre une forme juvénile, qui survient chez les enfants et les adolescents, et une forme adulte, dont souffrent les adultes.
La forme juvénile de déficit en alpha-N-acétylgalactosaminidase est appelée maladie de Schindler, tandis que la forme adulte est appelée maladie de Kanzaki. Mais il existe également des subdivisions entre la maladie de Schindler de type I pour la forme juvénile et la maladie de Schindler de type II pour la forme adulte. Un mélange des deux formes, connu sous le nom de maladie de Schindler de type III, est également possible.
Les trois formes de la maladie sont des oligosaccharidoses (glycoprotéinoses), qui comprennent la fucosidose et la maladie du stockage de l'acide sialique. Le nom de maladie de Schindler ou maladie de Schindler pour le déficit en alpha-N-acétylgalactosaminidase remonte au généticien allemand Detlev Schindler.
Il a été le premier à décrire la maladie en 1988. Un an plus tard, le médecin japonais T. Kanzaki a décrit la forme adulte. Il a découvert en 1991 qu'un déficit en alpha-N-acétylgalactosaminidase était la cause de la maladie.
causes
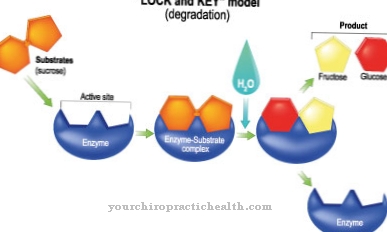
Le déficit en α-N-acétylgalactosaminidase est causé par une activité réduite de l'enzyme alpha-N-acétylgalactosaminidase. Des mutations faux ou absurdes sur le gène NAGA, qui code pour l'alpha-N-acétylgalactosaminidase, sont responsables du défaut enzymatique.
Le gène NAGA est situé sur le locus q11 du gène du chromosome 22. L'enzyme alpha-N-acétylgalactosaminidase a pour tâche de catalyser le clivage de la N-acétylgalactosamine à partir de divers glycolipides et glycoprotéines. Le défaut génétique entraîne une activité réduite de l'enzyme, de sorte que les substances qui ne sont pas métabolisées s'accumulent dans les cellules du corps.
Dans la plupart des cas, il s'agit de protéoglycanes, de glycosphingolipides et de glycoprotéines liées par N ou O. L'accumulation de ces derniers endommage la cellule et les organes affectés. Le déficit en alpha-N-acétylgalactosaminidase est si rare qu'il n'y a pas d'informations précises sur sa fréquence.
Jusqu'à présent, seuls douze patients de huit familles sont connus pour avoir développé la maladie de Schindler dans le monde. Les plaintes neurologiques ne se produisent pas chez tous les patients. Certains médecins soupçonnent l'influence de facteurs supplémentaires sur l'apparition de symptômes neurologiques. Cependant, il n'y a toujours aucune preuve pour étayer cette hypothèse.
Vous pouvez trouver votre médicament ici
➔ Médicaments contre la douleurSymptômes, maux et signes
Les symptômes d'un déficit en alpha-N-acétylgalactosaminidase dépendent de la forme particulière de la maladie. Dans le contexte de la maladie de Schindler de type I, une hypotonie musculaire progressive survient au cours de la première année de vie. De plus, les bébés atteints souffrent d'une régression psychomotrice prononcée.
Les perturbations de la séquence des mouvements, la tétraplégie spastique et les crises myocloniques cérébrales ont également été enregistrées comme symptômes. Il existe également un risque que l'enfant devienne aveugle. S'il existe une forme adulte de déficit en alpha-N-acétylgalactosaminidase, c'est-à-dire la maladie de Schindler de type 2, des symptômes similaires à la maladie de Fabry apparaissent.
Les patients ont des angiokératomes, qui sont des modifications cutanées bénignes. De nombreux patients présentent un léger retard mental. Une forme clinique intermédiaire est la maladie de Schindler de type III. Les personnes atteintes souffrent de dysfonctionnements neurologiques, de crises cérébrales et de troubles mentaux.
Cependant, des formes moins sévères sont également possibles, associées à des troubles psychiatriques et neurologiques légers, à un retard du développement du langage ou à des symptômes similaires à l'autisme.
Diagnostic et cours
Le diagnostic d'un déficit en alpha-N-acétylgalactosaminidase est possible grâce à la détection d'une activité NAGA réduite. A cet effet, des tests enzymatiques sont réalisés dans le plasma sanguin, les leucocytes ou les fibroblastes ou lymphoblastes en culture. La détection chromatographique des oligosaccharides dans l'urine permet également de diagnostiquer une carence enzymatique.
Une analyse ADN peut également être effectuée, mais ce n'est pas nécessaire dans la plupart des cas. Pendant la grossesse, le diagnostic prénatal du déficit enzymatique peut également être réalisé à l'aide d'une analyse de mutation du gène NAGA suite à un prélèvement de villosités choriales ou une amniocentèse. Cependant, la forme respective de la maladie ne peut être déterminée.
Le diagnostic différentiel du syndrome de Fabry, de la neurodégénérescence associée à la panthothénate kinase et de la dystrophie neuroaxonale infantile, qui présentent également des anomalies enzymatiques, est également important. L'évolution du déficit en alpha-N-acétylgalactosaminidase dépend de la forme respective de la maladie.
La maladie de Schindler de type I est considérée comme défavorable, tandis que le pronostic pour les deux autres types est plus favorable. Cependant, le nombre de malades est si petit qu'aucune déclaration significative ne peut être faite sur l'espérance de vie.
Complications
Les symptômes et les complications d'un déficit en A-N-acétylgalactosaminidase dépendent fortement de sa forme.Dans la plupart des cas, cependant, des troubles du mouvement surviennent, de sorte que les personnes touchées sont relativement limitées dans leur vie quotidienne. Des crampes se produisent également, qui sont associées à une douleur intense.
Les bébés en particulier souffrent de troubles du développement sévères, qui peuvent entraîner une spasticité chez l'enfant. Dans certains cas, le déficit en A-N-acétylgalactosaminidase conduit à une cécité complète chez le patient. La régression de l'intelligence conduit à un retard et donc à des handicaps.
Celles-ci restreignent généralement considérablement la vie quotidienne du patient et ont un effet négatif sur la qualité de vie. Les personnes touchées dépendent souvent de l'aide d'autres personnes. Le retard peut également conduire à un trouble de la recherche de mots ou à un trouble du langage. Il n'est pas possible de traiter le déficit en A-N-acétylgalactosaminidase de manière causale.
Pour cette raison, la personne affectée doit prendre des antibiotiques. Certaines complications doivent également être évitées directement. La pneumonie, par exemple, en fait partie. De plus, le patient peut également être dépendant d'une nutrition artificielle.
Quand devriez-vous aller chez le médecin?
Le déficit en A-N-acétylgalactosaminidase doit être définitivement traité par un médecin. Cette carence ne disparaît pas d'elle-même et il n'y a pas de guérison spontanée de cette maladie. Dans tous les cas, un médecin doit être consulté si les parents constatent une baisse des capacités motrices et mentales de leur enfant ou bébé. Les plaintes avec des mouvements ou des processus simples peuvent également indiquer un déficit en A-N-acétylgalactosaminidase et doivent être examinées par un médecin.
De plus, diverses spasticités sont également le signe d'une maladie. Il n'est pas rare que les patients souffrent de déficiences intellectuelles et d'autres dysfonctionnements. Si ces plaintes sont présentes, un traitement est absolument nécessaire. En l'absence de traitement, cela peut entraîner des plaintes et des complications considérables à l'âge adulte et ainsi réduire considérablement la qualité de vie de la personne touchée.
Le développement de la parole sévèrement retardé peut également être un signe de déficit en A-N-acétylgalactosaminidase et doit donc être étudié. En règle générale, un médecin généraliste peut être consulté pour déterminer le déficit en A-N-acétylgalactosaminidase. Le traitement ultérieur des plaintes individuelles est ensuite effectué par un spécialiste.
Médecins et thérapeutes dans votre région
Traitement et thérapie
Un traitement causal du déficit en alpha-N-acétylgalactosaminidase n'est pas envisageable. Par conséquent, l'approche médicale se limite au traitement des symptômes. Cela comprend un régime alimentaire raisonnable et un apport hydrique, la prévention des maladies infectieuses, ce qui peut également être fait en administrant des antibiotiques, ainsi que la réduction des crises cérébrales en administrant des médicaments antiépileptiques.
D'autres étapes de traitement comprennent des mesures de physiothérapie pour prévenir la pneumonie et les contractures, et pour réduire la douleur ou la spasticité à l'aide de médicaments. De plus, la prophylaxie par aspiration peut avoir lieu grâce à une alimentation artificielle.
Des études récentes ont montré que le déficit en α-N-acétylgalactosaminidase est un trouble de repliement des protéines, de sorte que la thérapie de remplacement enzymatique est considérée comme une mesure de traitement sensée. La thérapie génique est une autre approche thérapeutique envisageable pour le traitement des troubles du stockage lysosomal. Parce que la maladie de Schindler est héréditaire de manière autosomique récessive, un conseil génétique des familles touchées est recommandé.
Perspectives et prévisions
Le déficit en A-N-acétylgalactosaminidase peut entraîner différents symptômes. Dans la plupart des cas, cependant, la carence entraîne un retard du développement psychomoteur. À l'âge adulte, le patient est souvent dépendant de l'aide d'autres personnes et ne peut pas faire face seul à la vie quotidienne. Des troubles du mouvement surviennent également. Cela peut être remarqué par une boiterie ou par des troubles de la coordination.
Dans le pire des cas, le patient peut devenir aveugle ou souffrir de troubles visuels sévères dus au déficit en A-N-acétylgalactosaminidase. Il n'est pas rare que des déficiences intellectuelles se produisent et réduisent considérablement la qualité de vie de la personne concernée. Des troubles de la parole et des troubles de la recherche de mots surviennent également, ce qui peut affecter la vie. Les enfants en particulier sont affectés par l'intimidation et les taquineries en raison des symptômes d'un déficit en A-N-acétylgalactosaminidase.
Un traitement causal du déficit en A-N-acétylgalactosaminidase n'est pas possible, de sorte que le traitement vise principalement à réduire les symptômes. Diverses mesures thérapeutiques peuvent être utilisées pour réduire les troubles du développement. Dans de nombreux cas, cependant, le patient dépend d'une aide extérieure dans la vie quotidienne. L'espérance de vie n'est pas affectée par la carence.
Vous pouvez trouver votre médicament ici
➔ Médicaments contre la douleurla prévention
Il n'y a pas de mesures préventives connues contre un déficit en alpha-N-acétylgalactosaminidase. La maladie de Schindler est l'une des maladies les plus rares.
Suivi
Le déficit en A-N-acétylgalactosaminidase étant une maladie congénitale qui ne peut être traitée de manière causale mais seulement symptomatique, un traitement complet n'est pas possible. La personne touchée est dépendante d'un traitement à vie, c'est pourquoi les options de suivi sont très limitées.
En règle générale, en raison du déficit en A-N-acétylgalactosaminidase, le patient dépend souvent de la prise de médicaments et d'antibiotiques. Les interactions possibles avec d'autres médicaments doivent être prises en compte, bien que les antibiotiques ne doivent pas être pris avec de l'alcool. Si vous avez une crise, vous devez vous rendre à l'hôpital ou appeler directement un médecin urgentiste.
De plus, les poumons doivent être épargnés pour éviter l'inflammation. Par conséquent, les patients présentant un déficit en A-N-acétylgalactosaminidase ne doivent en aucun cas fumer. Une alimentation saine et un mode de vie généralement sain ont un effet très positif sur l'évolution de la maladie.
Si le patient souhaite avoir des enfants, un conseil génétique est utile pour éviter que la maladie ne se transmette aux enfants. Étant donné que les patients souffrent souvent d'une mobilité réduite, la physiothérapie est utile pour atténuer cela. De nombreux exercices peuvent également être pratiqués chez vous.
Tu peux le faire toi-même
Un déficit existant en A-N-acétylgalactosaminidase est une maladie incurable. Un traitement médical est donc indispensable. Les mesures d'auto-traitement ne peuvent être basées que sur les symptômes pour faciliter la vie quotidienne. Il est important que les parents observent l'enfant de près.
Les parents peuvent mieux aider leur enfant par la compréhension et la patience, l'amour et l'attention. De plus, les traitements d'ergothérapie et d'orthophonie reposent toujours sur un double concept: la thérapie sur place avec le spécialiste et la réalisation des exercices à domicile. Les parents doivent être actifs ici de manière cohérente et avec patience. Une alimentation riche en vitamines et minéraux, une activité physique régulière et beaucoup d'air frais doivent être assurés. Cela renforce le système immunitaire et réduit le risque d'infection. Si la personne a tendance à saisir, elle ne doit pas être seule pendant une longue période et elle doit être vérifiée pour voir si elle peut se blesser en cas de crise.
La plupart du temps, les patients ne peuvent pas faire face eux-mêmes à la vie quotidienne et nécessitent des soins constants. Si les parents ne peuvent pas se le permettre - en particulier avec les enfants plus âgés et les adultes - ils ne devraient pas avoir peur de demander de l'aide. Cela peut prendre la forme d'une infirmière ou d'un placement dans un établissement adéquat. Si vous souhaitez toujours avoir des enfants, une consultation avec un spécialiste est recommandée, car la maladie est héréditaire.

.jpg)