le Syndrome de Bardet-Biedl, aussi Syndrome de Laurence-Moon-Biedl-Bardet (LMBBS), est une maladie du domaine des ciliopathies qui survient exclusivement en raison de l'hérédité. Le syndrome se manifeste sous la forme de multiples malformations déclenchées par des changements (mutations) sur différents emplacements de gènes ou chromosomes.
Qu'est-ce que le syndrome de Bardet-Biedl?

© Creativa Images - stock.adobe.com
Le tableau clinique défini par les médecins Moon et Laurence et plus tard par Bardet et Biedl est une maladie dans laquelle la dystrophie rétinienne apparaît comme une caractéristique médicalement significative en combinaison avec d'autres symptômes. En raison de cette situation médicale initiale compliquée, la détermination finale de la maladie BBS est difficile. Ce tableau clinique a été enregistré médicalement pour la première fois en 1866.
Quatre personnes examinées avaient une rétinite pigmentaire (dystrophie rétinienne, RP) associée à une paraplégie (paralysie spastique) ainsi qu'un hypogénitalisme (organes génitaux sous-développés) et un handicap mental. En 1920, le médecin français Bardet décrivit une maladie composée de RP (dystrophie rétinienne), d'hypogénitalisme, de polydactylie et d'obésité.
Le pathologiste pragois Biedl a également constaté une débilité (confusion mentale). En 1925, les chercheurs Weiss et Solis-Cohen ont résumé les cas connus et décrit le tableau clinique comme Syndrome de Laurence-Moon-Biedl-Bardet.
causes
Dans les années qui ont suivi, la littérature médicale a de plus en plus souligné que les cas enregistrés par Laurence et Moon sont une forme spéciale rare qui ne se produit que dans des cas isolés avec BBS. Des résultats de recherche médicale plus récents attribuent le syndrome de Bardet-Biedl au domaine des ciliopathies (maladies ciliaires).
Ces maladies montrent un dysfonctionnement commun des soi-disant cils (petits processus, antennes), qui surviennent sur la majorité des cellules de l'organisme humain. Les ciliopathies sont caractérisées par des transitions fluides et des chevauchements entre différentes maladies ciliaires.
Symptômes, maux et signes
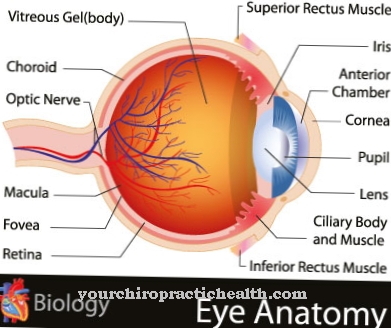
La principale caractéristique de la dystrophie rétinienne héréditaire est un terme générique qui décrit le début de la perte de fonction et la dégénérescence (destruction) ultérieure des photorécepteurs. Ils conduisent à une perte progressive (progressive) de la fonction visuelle. Les troubles visuels à évolution rapide apparaissent généralement très tôt chez les enfants entre quatre et dix ans. Ils se font sentir de différentes manières, en fonction des photorécepteurs affectés.
En forme de "bâtonnet-cône" avec l'évolution caractéristique de la rétinite pigmentaire (RP), la maladie trouve son origine dans la périphérie rétinienne (rétine externe) et évolue vers une dégénérescence maculaire (destruction de la vue nette) via une perte progressive du champ de vision.
Avec l'obésité (obésité), le corps présente une accumulation pathologique de tissu adipeux. Dans le cas du BBS, des accumulations anormalement accrues de graisse sur les jambes, le ventre, les fesses, les bras, la poitrine et les hanches se produisent principalement sous forme d'obésité du tronc, le tronc, les jambes et les cuisses étant particulièrement affectés. La polydactylie est un symptôme notable et une caractéristique importante du syndrome de Bardet-Biedl. Le constat n'est pas facile car la polydactylie rudimentaire est corrigée chirurgicalement après la naissance.
Les rayons X peuvent fournir des informations supplémentaires. La polydactylie peut apparaître avec différents signes, par exemple comme un orteil ou un appendice de doigt rudimentaire. Un orteil ou un doigt peut être formé en plus ou seulement partiellement. L'hexadactylie unilatérale sur le pied et / ou la main a un lien supplémentaire, l'hexadactylie bilatérale se produit sur les deux pieds et / ou les mains.
Les orteils ou les doigts qui se sont développés ensemble (syndactylie) et le raccourcissement d'un ou plusieurs orteils ou doigts (brachydactylie) sont également des signes de BBS. Seuls quelques patients ont les quatre extrémités touchées. Le retard du développement mental est différent. Seul un petit nombre de personnes atteintes présentent un retard mental sévère. Une intelligence normalement formée est possible.
Les enfants apprennent à parler et à marcher tard, et présentent parfois des problèmes de comportement tels que des troubles anxieux. Les comportements compulsifs ou autistiques, un seuil bas de frustration et une émotivité instable sont d'autres effets secondaires possibles. Le familier est préféré, mais les modifications sont rejetées. Les anomalies des organes génitaux internes et externes sont fréquentes.
D'autres changements sont l'hypospadias (l'ouverture de l'urètre est au-dessus ou au-dessous, au lieu de l'avant du pénis), l'abdomen ou les testicules inguinaux, les constrictions urétrales, la constriction du prépuce et les valves urétrales postérieures. Chez les patientes, une atrésie vaginale (le vagin n'est pas ouvert), des ouvertures urétrales manquantes et des lèvres internes réduites sont connues.
Il n'est pas rare que les femmes touchées aient des cycles menstruels irréguliers. Les modifications rénales sont des effets secondaires courants. Le résultat dépend de l'examen des voies urinaires inférieures et des reins par échographie (échographie).
Diagnostic et évolution de la maladie
Le syndrome de Bardet-Biedl (BBS) a six symptômes principaux, mais ils ne surviennent pas ensemble dans tous les cas. Les médecins supposent un résultat correspondant si au moins quatre des principaux symptômes sont présents. Alternativement, il y a une forte probabilité que la maladie soit présente si le patient présente trois symptômes principaux et deux symptômes secondaires.
Les six principaux symptômes sont la dystrophie rétinienne, l'obésité (accumulation anormale de tissu adipeux, le surpoids), la polydactylie (excès d'orteils et / ou de doigts), le retard mental (retard de développement mental), l'hypogénitalisme (organes génitaux sous-développés) et les maladies rénales. Les symptômes secondaires de basse fréquence comprennent des retards d'élocution, des déficits d'élocution, des malformations cardiaques, l'ataxie (troubles de la coordination des mouvements), l'asthme, le diabète sucré (diabète), la maladie de Crohn (inflammation du gros et / ou de l'intestin grêle), la dysplasie des côtes et des vertèbres et la cyphoscoliose (anémie vertébrale) sur.
Complications
Avec le syndrome de Laurence-Moon-Biedl-Bardet, les personnes atteintes souffrent généralement d'une perte de la fonction visuelle. La perte ne se produit pas soudainement, mais progressivement. Dans le pire des cas, les personnes touchées deviendront complètement aveugles, ce qui ne peut généralement plus être traité.
Surtout chez les jeunes et les enfants, la cécité peut entraîner de graves troubles psychologiques, voire une dépression. Les patients sont clairement limités dans leur vie quotidienne et souffrent d'un champ de vision très réduit. Dans de nombreux cas, le syndrome de Laurence-Moon-Biedl-Bardet conduit également à des problèmes de comportement, de sorte que les enfants en particulier peuvent souffrir d'intimidation ou de taquineries.
Le développement des enfants est également considérablement retardé et restreint par le syndrome. Des troubles anxieux peuvent également survenir. Il n'est pas rare que le syndrome de Laurence-Moon-Biedl-Bardet entraîne des plaintes psychologiques et une dépression chez les proches ou les parents. Un traitement causal du syndrome de Laurence-Moon-Biedl-Bardet n'est malheureusement pas possible.
Certaines plaintes peuvent être limitées. Une évolution totalement positive de la maladie ne s'installe cependant pas. Le syndrome ne réduit pas l'espérance de vie du patient. Dans certains cas, les personnes touchées ont parfois besoin de l'aide d'autres personnes dans leur vie quotidienne.
Quand devriez-vous aller chez le médecin?
Le syndrome de Laurence-Moon-Biedl-Bardet étant une maladie héréditaire, le diagnostic peut être posé dans l'utérus. Au plus tard après la naissance, un médecin doit être consulté si des symptômes typiques tels que des troubles visuels ou l'obésité sont constatés. Les malformations des orteils et des doigts sont également un indicateur clair d'une maladie.Les parents qui remarquent des symptômes chez leur enfant doivent en informer immédiatement le pédiatre.
Un examen complet fournit des informations sur la maladie. Par la suite, la thérapie est généralement initiée directement, qui consiste en divers traitements par des orthopédistes, des neurologues, des ophtalmologistes, des internistes et des thérapeutes ainsi que des physiothérapeutes. D'autres visites chez le médecin sont nécessaires si le traitement n'a pas l'effet souhaité. Un avis médical est également requis dans les situations d'urgence, par exemple si l'enfant tombe à la suite d'une malformation ou fait soudainement une crise. Si la personne malade montre des signes d'inconfort émotionnel, les parents doivent consulter un thérapeute approprié. Les enfants plus âgés peuvent contacter le psychologue scolaire avec leurs parents et discuter des mesures appropriées.
Thérapie et traitement
Cette maladie survient sur la base de l'hérédité autosomique récessive, ce qui signifie que les deux copies (allèles) d'un gène BBS présentent un changement (mutation). Les parents du patient sont «de sang-mêlé» et portent chacun un allèle modifié et un allèle inchangé du gène correspondant. Ils n'ont pas la maladie. Les enfants ne tombent malades que si leur père et leur mère transmettent l'allèle muté. Avec des enfants supplémentaires, la probabilité de redoublement est de 25%.
Une option thérapeutique causale n'est pas encore connue, car certains symptômes de la maladie ne peuvent pas encore être attribués de manière concluante aux divers changements génétiques. Les symptômes et leurs manifestations apparaissent différemment même chez les frères et sœurs malades. Étant donné que le tableau complet caractéristique du BBS n'est présent que dans de rares cas, en particulier chez les jeunes enfants, un diagnostic correspondant est difficile.
En raison des symptômes oligosymptomatiques fréquemment présents, avec lesquels apparaissent très peu de symptômes atypiques et seulement légèrement prononcés, d'autres tableaux cliniques possibles doivent être pris en compte dans le diagnostic différentiel. Des modifications d'un même gène peuvent conduire à des tableaux cliniques différents, par exemple le syndrome de Joubert, Bardet-Biedl ou Meckel-Gruber.
Perspectives et prévisions
Le pronostic de la présence du syndrome de Laurence-Moon-Biedl-Bardet est généralement mauvais car les multiples malformations sont congénitales et incurables. Si quatre des six principaux symptômes apparaissent, le diagnostic de syndrome de Laurence-Moon-Biedl-Bardet est confirmé. De nombreux symptômes secondaires s'ajoutent aux principaux symptômes. Cela inclut une cécité rampante.
C'est en raison de la complexité des symptômes qu'il n'y a pas de perspective de guérison. Il n'y a qu'une chance médiocre de soulagement notable des symptômes. Le nombre de malformations et de troubles possibles du syndrome de Laurence-Moon-Biedl-Bardet est si grand que la maladie héréditaire est difficile à traiter. Dans tous les cas, l'évolution de cette maladie génétique ne peut être influencée. Cependant, les symptômes actuels peuvent être partiellement atténués.
Cependant, le mauvais pronostic global ne réduit pas l'espérance de vie des personnes touchées. À un âge avancé et après être devenus aveugles, les personnes touchées peuvent être en permanence dépendantes de l'aide ou des soins. Grâce à des efforts médicaux interdisciplinaires, de nombreuses personnes atteintes du syndrome de Laurence-Moon-Biedl-Bardet peuvent connaître une évolution un peu plus douce de la maladie.
Les problèmes visuels croissants représentent une partie difficile à traiter et problématique de la maladie. Des déficiences visuelles croissantes se produisent déjà chez les jeunes enfants atteints. Ils s'aggravent avec le temps. Les problèmes de vision ne doivent pas conduire à la cécité chez toutes les personnes touchées. Les séquelles psychologiques du syndrome de Laurence-Moon-Biedl-Bardet peuvent généralement être bien traitées.
la prévention
La prévention dans le sens de la prévention de cette maladie n'est pas possible. Une surveillance régulière des symptômes et des symptômes qui les accompagnent est importante. Des contrôles répétés de la pression artérielle et de la fonction rénale, des conseils nutritionnels, la physiothérapie et l'ergothérapie ainsi que l'orthophonie sont des approches thérapeutiques possibles.
Suivi
Dans la plupart des cas, les personnes atteintes du syndrome de Laurence-Moon-Biedl-Bardet n'ont pas d'options de suivi spéciales disponibles, de sorte qu'un médecin doit être contacté et consulté très tôt dans cette maladie. En règle générale, l'auto-guérison ne peut pas se produire, un traitement par un médecin est donc toujours nécessaire.
Le syndrome de Laurence-Moon-Biedl-Bardet étant une maladie héréditaire, la personne concernée doit subir un examen génétique et des conseils si elle souhaite avoir des enfants afin que le syndrome de Laurence-Moon-Biedl-Bardet ne passe pas à sa progéniture est transmis. Dans de nombreux cas, les personnes touchées dépendent d'interventions chirurgicales pour atténuer les malformations et les déformations.
Ici, la personne touchée doit absolument se reposer après la procédure et prendre soin de son corps. L'effort ou d'autres activités physiques et stressantes doivent être évités dans tous les cas afin de ne pas alourdir inutilement le corps. Le syndrome de Laurence-Moon-Biedl-Bardet pouvant également conduire à un comportement anormal, les parents doivent soutenir et encourager l'enfant dans son développement. Des discussions aimantes et intensives avec l'enfant sont également nécessaires pour éviter les troubles psychologiques ou la dépression.
Tu peux le faire toi-même
Le syndrome de Laurence-Moon-Biedl-Bardet présente divers symptômes, le patient souffrant souvent le plus d'une fonction visuelle altérée. Même avec les enfants, la capacité habituelle de voir commence à se détériorer, de sorte que ce sont les parents qui présentent l'enfant à un médecin et accélèrent ainsi le diagnostic. De cette manière, la maladie peut être traitée rapidement, bien que les options de traitement n'aient jusqu'à présent été que de nature symptomatique.
Les troubles visuels augmentent de plus en plus chez les enfants malades et altèrent ainsi considérablement la vie quotidienne, de sorte que la qualité de vie des personnes atteintes diminue. Car les problèmes de vision développent de nombreuses difficultés pour le patient lorsqu'il fréquente l'école, dans son temps libre et au regard de son intégrité physique. Le risque d'accidents augmente également de manière significative, par exemple dans le trafic routier. C'est pourquoi les parents accompagnent leurs enfants malades dans la mesure du possible ou engagent du personnel infirmier afin que le patient ne soit pas laissé à lui-même.
Dans certains cas, la maladie se propage jusqu'à la cécité. Puisqu'une telle évolution est déjà évidente à l'avance, les patients s'y préparent. Les parents réaménagent l'espace de vie afin qu'il ne contienne aucune source de danger pour la personne malvoyante. De plus, les victimes aveugles apprennent à utiliser un long bâton pour pouvoir se déplacer de manière autonome en dehors de leur propre maison.
.jpg)

.jpg)


.jpg)

.jpg)







.jpg)