Il y en a 45 différents au total maladies du stockage lysosomal, qui constituent un groupe hétérogène de maladies métaboliques congénitales. Les personnes qui souffrent de l'une de ces maladies ont un défaut génétique. Toutes les maladies de stockage ont une chose en commun: une certaine enzyme est soit absente, soit partiellement fonctionnelle.
Qu'est-ce que la maladie du stockage lysosomal?

© designua - stock.adobe.com
Ces maladies de stockage congénitales sont rares, car moins de cinq personnes sur 10000 sont touchées. Les diverses maladies ont une évolution très différente et les symptômes peuvent varier considérablement.
Les formes les plus connues de maladie du stockage lysosomal sont la maladie de Fabry, la maladie de Gaucher, la maladie de Pompe et la mucopolysaccharidose (MPS). Ils sont souvent appelés «orphelins de la médecine» car le chemin vers un diagnostic spécifique et une thérapie adaptée peut être très long. Parfois, les personnes touchées peuvent mettre des années à découvrir ce qui leur arrive.
causes
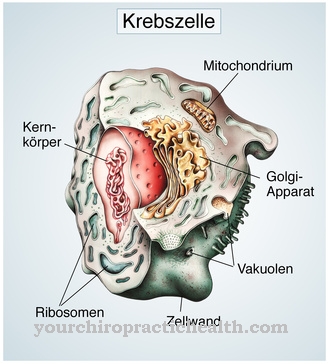
Les maladies du stockage lysosomal sont caractérisées par certaines formes de maladies métaboliques héréditaires. Le patient n'a pas une enzyme importante qui assure le bon fonctionnement de l'équilibre métabolique. Dans la forme la moins prononcée, cette enzyme n'est au moins pas présente en quantité suffisante.
La tâche des enzymes est d'éliminer les polluants et les déchets qui s'accumulent dans l'organisme humain via le métabolisme via les lysosomes, ou de les traiter à nouveau de manière à ce que les symptômes n'apparaissent pas.
En cas de carence enzymatique, ce cycle d'élimination fonctionnant sans heurts n'est plus garanti. Les substances nocives se déposent dans les cellules et perturbent le cycle métabolique. Dans la phase initiale, les perturbations n'ont pas d'effet notable, il n'y a que quelques restrictions. Cependant, si ce trouble métabolique n'est pas traité en raison d'un déficit enzymatique, les symptômes se multiplient car les cellules deviennent très grossies.
Symptômes, maux et signes
Dans le pire des cas, ils font faillite. Les conséquences sont des dommages aux os, au système nerveux, à la rate, aux reins, aux muscles ou au cœur. En raison de l'activité enzymatique réduite ou absente, la maladie de Fabry entraîne le stockage des graisses (globotriaosylcéramide, Gb3) dans les cellules. Ces dépôts indésirables peuvent entraîner une douleur intense dans les orteils ou les doigts, un accident vasculaire cérébral et des lésions rénales.
Diagnostic et évolution de la maladie
Ce tableau clinique affecte en même temps différents systèmes: vaisseaux sanguins, reins, cœur et système nerveux. La maladie de Gaucher autosomique récessive provoque une mutation de l'enzyme «bêta-glucocérébrosidase» et conduit à une accumulation de substrat au sein des cellules, en particulier dans les macrophages (phagocytes) appartenant au système réticulo-endothélial. La formule sanguine change, le foie et la rate sont hypertrophiés et les os font mal.
La maladie est progressive et est principalement ethnique, car elle survient dans la plupart des cas chez des personnes d'origine juive. La maladie de Pompe est également connue sous le nom de «déficit en maltase acide». Le tableau clinique appartient au groupe de la glycogénèse de type II Les personnes atteintes sont dépourvues de l'enzyme "alpha-1,4-glucosidase" (acide maltase) ou n'est pas disponible en quantité suffisante. En raison de la dégradation du glycogène dans les muscles, les patients souffrent de la destruction des cellules musculaires sous forme de stockage de sucre.
La mucopolysaccharidose de type I (MPS), également connue sous le nom de maladie de Hunter, a diverses causes cliniques. La maladie de Hurler est la forme la plus sévère et la maladie de Scheie est à la fin de la pathogenèse clinique. Il existe des transitions de caractéristiques différentes entre ces deux formes de progression. La propriété la plus frappante est la dégradation altérée des glucides qui s'accumulent dans les lysosomes des cellules.
Les patients atteints de la maladie de Hunter peuvent présenter une petite taille, une rate et un foie hypertrophiés, des traits macroscopiques, une peau épaissie, une langue élargie et des difficultés respiratoires. De plus, le squelette est souvent modifié au niveau du bassin, de la colonne vertébrale, des os de la main et du crâne. Des hernies ombilicales et [[hernies inguinales] sont possibles.
Complications
Dans la plupart des cas, les symptômes ou complications apparaissent très tardivement dans cette maladie. Pour cette raison, il est diagnostiqué tardivement, ce qui rend le traitement précoce dans la plupart des cas impossible. Sans traitement, diverses plaintes et lésions des organes internes surviennent au fur et à mesure que la maladie progresse.
Les reins, le foie et la rate sont particulièrement touchés. Le cœur peut également être affecté par cette maladie, qui peut entraîner la mort cardiaque dans le pire des cas. De plus, des lésions rénales se produisent et les personnes touchées souffrent souvent de douleurs aux orteils ou aux doigts. La paralysie peut également survenir si le cerveau a été endommagé par cette maladie. Le foie et la rate peuvent être hypertrophiés et provoquer une douleur intense.
Il n'est pas rare que les os de la personne touchée soient cassants et douloureux. Le traitement de cette maladie s'avère difficile. Dans de nombreux cas, l'espérance de vie de la personne touchée est considérablement réduite. Il n'y a généralement pas de complications particulières lors de l'utilisation de médicaments. Cependant, une évolution positive de la maladie ne peut être garantie dans tous les cas.
Vous pouvez trouver votre médicament ici
➔ Médicaments contre la douleurQuand devriez-vous aller chez le médecin?
La perte de cheveux, les problèmes articulaires et les troubles des organes sont des signes possibles d'une maladie du stockage lysosomal. Une visite chez le médecin est recommandée si les symptômes persistent ou apparaissent soudainement sans qu'une cause soit trouvée. Si les symptômes sont liés à un défaut enzymatique déjà diagnostiqué ou à une autre maladie grave, le médecin responsable doit être consulté. Une maladie de stockage non traitée peut entraîner une démence, une infertilité, des neuropathies et d'autres complications, dont certaines mettent la vie en danger. Par conséquent, tous les symptômes imaginables doivent être examinés, même s'il n'y a pas de suspicion spécifique.
Les symptômes de la maladie du stockage lysosomal peuvent apparaître par étapes ou se développer insidieusement, mais nécessitent toujours un examen et un traitement. Il est préférable que les personnes concernées parlent directement à leur médecin de famille ou à un interniste. La thérapie proprement dite a généralement lieu dans une clinique spécialisée pour les maladies internes, bien que la physiothérapie ou la psychothérapie puissent être associées en fonction des symptômes. En particulier, des mesures thérapeutiques sont indiquées en raison de l'évolution souvent négative de la maladie.
Thérapie et traitement
Selon la rapidité avec laquelle un diagnostic adéquat est posé, ces maladies héréditaires peuvent être très bien traitées avec une thérapie enzymatique substitutive, de sorte que les personnes touchées ont beaucoup moins de plaintes et donc une meilleure qualité de vie. Cette thérapie de remplacement est utilisée selon le tableau clinique.
Les personnes atteintes de la maladie de Gaucher sont dépourvues de «l'enzyme ß-glucocérébrosidase», qui est produite par biotechnologie et infusée dans le corps du patient. Les lysosomes agissent efficacement et sont capables d'absorber des substances de leur environnement immédiat. Pour cette raison, les enzymes artificiellement utilisées sont modifiées de manière à pouvoir être fournies aux lysosomes de manière idéale.
Les macrophages (phagocytes) décomposent les glucocérébrosides qui se sont accumulés dans les cellules. Cette thérapie peut être comparée à une insulinothérapie pour le diabète sucré, à la différence qu'il ne s'agit pas d'une hormone manquante mais d'une enzyme qui n'est pas fournie. Le corps décompose régulièrement toutes les substances, y compris l'enzyme artificielle fournie.
En raison de cette dégradation régulière de la substance, les patients doivent suivre cette thérapie par perfusion régulièrement jusqu'à la fin de leur vie. La thérapie enzymatique substitutive n'agit pas de manière symptomatique, mais combat directement la cause de la maladie héréditaire. Les médecins appellent cette thérapie causale. Les principes de la thérapie doivent être utilisés pour les quatre maladies de stockage courantes mentionnées ci-dessus.
Les patients sous Pompe sont également traités par perfusion. Dans cette maladie, l'enzyme inexistante "acide alpha glucosidase" est fournie et aide à décomposer le glycogène qui s'est accumulé dans les lysosomes des muscles. Chez les patients atteints de la maladie de type «mucopolysaccharidose de type I», l'enzyme lysosomale «alpha-iduronidase» n'est pas présente ou n'est pas présente en quantité suffisante. C'est l'une des maladies de stockage les plus rares dans laquelle les molécules de sucre s'accumulent dans les organes et les tissus.
Si le processus est normal, l'enzyme décompose les mucopolysaccharides. Les molécules de sucre sont à longue chaîne et sont impliquées dans le développement des tissus de soutien et conjonctifs, par exemple les os, la peau, les fluides articulaires et le cartilage. Si le cours normal de la dégradation est perturbé en raison du manque d'enzyme, les glycosaminoglycanes pathologiques (GAG) s'accumulent dans les cellules individuelles. Les futures options thérapeutiques visent à prendre des comprimés.
Perspectives et prévisions
Le pronostic de la maladie de stockage est mauvais. Une disposition génétique s'est avérée être la cause du trouble de santé. Les exigences légales interdisent aux médecins et aux scientifiques de modifier la génétique humaine. Pour cette raison, la maladie dure toute la vie et n'a aucune perspective de guérison.
Le médecin traitant se concentre sur le traitement des symptômes qui surviennent. Si elles ne sont pas traitées, diverses plaintes augmenteront avec le temps. Le système osseux est endommagé et des problèmes d'organes surviennent. Dans le pire des cas, les organes internes fonctionneront mal et finalement leur fonction échouera. Cela menace la personne concernée de mort prématurée.
Le défi de la maladie réside dans le diagnostic. Chez un grand nombre de patients, les plaintes notables et fortement perceptibles n'apparaissent que plus tard dans la vie. En conséquence, le trouble génétique reste longtemps inaperçu et le traitement précoce de la maladie est difficile. Plus un diagnostic est posé tardivement, plus l'évolution est défavorable. À un stade avancé de la maladie, les organes internes ou les articulations sont déjà gravement endommagés. Des interventions chirurgicales sont nécessaires et si la maladie évolue de manière défavorable, un seul organe donneur peut sauver la vie de la personne touchée. Un traitement précoce est donc essentiel pour un meilleur pronostic.
la prévention
Puisqu'il s'agit d'un défaut génétique congénital qui empêche l'expression d'une enzyme, cette maladie ne peut être traitée de manière préventive. Cependant, les dernières réalisations du génie génétique pourraient fournir une approche dans ce domaine.
Suivi
Avec cette maladie, les gens souffrent d'un certain nombre de complications et d'affections différentes. En règle générale, tout cela a un effet très négatif sur la qualité de vie de la personne touchée, de sorte qu'un diagnostic doit être posé très tôt.Plus tôt un médecin est consulté, meilleure est généralement l'évolution de cette maladie.
La gravité de cette maladie peut varier considérablement, de sorte qu'une prédiction générale n'est souvent pas possible. Les personnes touchées souffrent de graves dommages aux organes internes. Les reins et le cœur sont principalement affectés, de sorte que l'enfant peut mourir dans les premiers jours si les symptômes ne sont pas corrigés à temps. Il existe également des dépôts de graisse dans différentes parties du corps.
Les doigts et les orteils sont particulièrement touchés, ce qui peut conduire à une esthétique considérablement réduite pour la personne touchée. En règle générale, les reins et le cerveau sont endommagés ultérieurement, de sorte que la personne touchée meurt à la suite de ces dommages. Les parents et les proches souffrent également souvent de dépression ou d'autres troubles mentaux dus à la maladie.
Tu peux le faire toi-même
Les maladies du stockage lysosomal nécessitent très souvent des soins médicaux intensifs. Souvent, les opportunités d'auto-assistance sont insuffisantes. Les parents des enfants affectés vivent souvent un stress sévère dans leur environnement familial parce que leur enfant a besoin de soins et d'attention constants.
Les tableaux cliniques des différentes maladies de stockage sont différents. Il existe des formes à la fois faciles et très difficiles. La maladie de Gaucher en est un exemple. L'aide des parents se limite souvent à nourrir l'enfant gravement handicapé. Dans les cas plus légers, l'espérance de vie peut être presque normale. Néanmoins, une surveillance médicale constante est nécessaire pour éviter d'éventuelles complications. L'activité physique régulière fait partie des thérapies d'accompagnement qui peuvent également être pratiquées à domicile. De plus, un examen approfondi de dépistage du cancer doit être organisé. Cela nécessite des visites constantes chez le médecin avec leur enfant des parents. Il en va de même pour les autres maladies du stockage lysosomal.
Dans le cas de certaines maladies, en plus des handicaps physiques, des déficiences mentales peuvent également survenir, qui nécessitent encore un soutien particulier. Dans les formes bénignes de certaines maladies, telles que la maladie de Hunter, seuls des changements squelettiques et une dysmorphie faciale se produisent initialement. Ici, cependant, le patient atteint est souvent capable de mener une vie indépendante. Cependant, des examens médicaux constants sont également nécessaires ici afin d'exclure d'éventuelles complications telles que l'insuffisance cardiaque ou les maladies respiratoires. Le patient peut faire face au stress psychologique causé par des déformations physiques grâce à des conseils psychologiques.
.jpg)
.jpg)






.jpg)



.jpg)

.jpg)