le Syndrome de Meckel-Gruber (MKS) est une maladie héréditaire. Elle se caractérise par de graves incapacités congénitales. Les nouveau-nés atteints meurent généralement dans les deux premières semaines suivant la naissance.
Qu'est-ce que le syndrome de Meckel-Gruber?

© hywards - stock.adobe.com
le Syndrome de Meckel-Gruber est une maladie héréditaire caractérisée par des kystes rénaux, des troubles du développement et des troubles du système nerveux central. La maladie est également sous le nom Syndrome de Meckel connu.
En Allemagne, 0,7 à 7,5 nouveau-nés pour 100 000 naissances sont statistiquement touchés par la maladie. La maladie est beaucoup plus courante en Finlande. Un nouveau-né sur 9000 est touché ici. S'il n'y a pas d'interruption de grossesse, les enfants meurent souvent pendant la période périnatale, c'est-à-dire avant le septième jour de la vie.
causes
La maladie est héréditaire par un défaut génétique autosomique récessif. Dans un mode d'hérédité autosomique récessif, le défaut génétique se situe sur l'une des 22 paires dites d'autosomes. Les autosomes sont des chromosomes qui, contrairement aux gonosomes, n'ont aucune influence sur le sexe. Le syndrome de Meckel-Gruber se transmet quel que soit le sexe. Récessif signifie que la maladie n'éclate que lorsque deux gènes malades, l'un du père et l'autre de la mère, sont transmis à l'enfant.
Pour qu'un enfant développe le syndrome de Meckel-Gruber, le père et la mère de l'enfant doivent être porteurs de la maladie. Les parents ne présentent aucun symptôme car ils ne portent chacun qu'un seul gène malade. Le deuxième gène malade est absent pour l'apparition de la maladie. Les parents sont également appelés conducteurs, c'est-à-dire porteurs du gène défectueux. Si les deux parents sont conducteurs, la probabilité de développer le syndrome de Meckel-Gruber est statistiquement de 25 pour cent.
Si les parents sont apparentés, la probabilité augmente. Le gène responsable de la maladie n'a jusqu'à présent été que partiellement retrouvé. Il semble que des changements dans trois emplacements de gènes différents soient responsables de la maladie. Ils sont sur les chromosomes 17, 11 et 8.
Symptômes, maux et signes
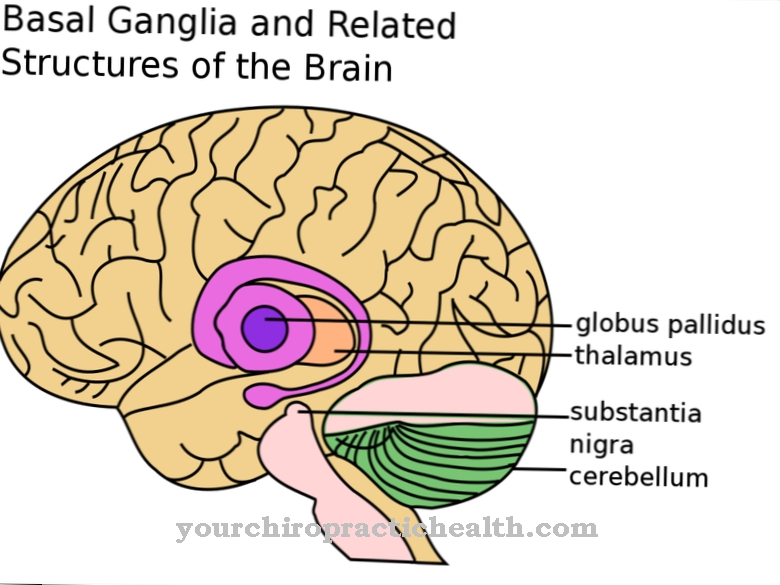
Les reins kystiques sont caractéristiques du syndrome de Meckel-Gruber. De nombreuses vésicules remplies de liquide se forment dans les reins, de sorte que la fonction de filtre des reins est sévèrement limitée. La formation de kystes rénaux est obligatoire, c'est-à-dire que s'il n'y a pas de kystes rénaux, il ne peut pas être question de syndrome de Meckel-Gruber. Des kystes hépatiques peuvent également survenir. Ceux-ci conduisent parfois à une fibrose hépatique. Les enfants souffrent également d'encéphalocèle.
Le cerveau est mal conçu et le crâne n'est souvent pas correctement fermé, de sorte que des parties du cerveau se gonflent hors du crâne. D'autres malformations cérébrales ont été observées. Une fente labiale et palatine, une malformation de la zone buccale, peut également survenir dans le syndrome de Meckel-Gruber. Souvent, les nouveau-nés souffrent également de microphtalmie. Avec la microphtalmie, les yeux sont inhabituellement petits ou peut-être seulement rudimentaires.
Un autre symptôme du syndrome de Meckel-Gruber est la soi-disant polydactylie, les multiples doigts. Il y a donc plus de dix doigts ou dix orteils. Un double pouce des deux côtés est particulièrement fréquent, de sorte que les malades ont douze doigts au lieu de dix doigts.
Un situs inversus est également un phénomène de la maladie héréditaire. Tous les organes sont inversés en miroir de l'autre côté du corps. Par exemple, le cœur est à gauche et le foie à droite. D'autres symptômes du syndrome de Meckel-Gruber sont des malformations des voies biliaires et des poumons sous-développés.
Diagnostic et évolution de la maladie
Le rein à kyste est un indice important dans le diagnostic du syndrome de Meckel-Gruber. Les critères diagnostiques minimaux de la fièvre aphteuse sont les modifications kystiques du rein, les modifications fibrotiques du foie et l'encéphalocèle ou d'autres malformations du système nerveux central. Le diagnostic prénatal de la maladie est effectué par échographie.
Un fœtus malade a un intérieur du crâne kystiquement altéré, et parfois d'autres défauts du crâne. Les reins sont également hypertrophiés. Ces indications du syndrome de Meckel-Gruber se retrouvent déjà à la fin du premier trimestre de grossesse. Au fur et à mesure que la grossesse se poursuit, d'autres anomalies peuvent être détectées par échographie.
Un test de liquide amniotique révèle une augmentation du taux d'alpha-foetoprotéine. Ceci est causé par des anomalies du crâne et du SNC et est un signe certain d'une déformation grave du crâne.
Complications
Malheureusement, en raison du syndrome de Meckel-Gruber, dans la plupart des cas, la patiente meurt quelques semaines après la naissance. Pour cette raison, les proches et les parents de l'enfant en particulier sont affectés par des troubles physiques graves ou une dépression et ont donc également besoin d'un traitement psychologique. Les personnes touchées souffrent elles-mêmes de graves handicaps dus au syndrome de Meckel-Gruber et ne peuvent survivre pour cette raison.
Surtout, cela conduit à des malformations des reins et du foie du patient, entraînant une insuffisance et donc la mort. Les patients souffrent également d'une soi-disant fente palatine et donc de restrictions à la prise de nourriture. L'espérance de vie des patients est honnêtement limitée et réduite par le syndrome de Meckel-Gruber.
Malheureusement, il n'est pas possible de traiter le syndrome de Meckel-Gruber ou de résoudre les symptômes. Les enfants meurent très tôt après la naissance. Malheureusement, d'autres mesures pour soutenir la vie ne sont pas possibles, de sorte qu'il n'y a pas d'autres complications. En règle générale, les parents ont besoin d'un traitement psychologique après le décès de l'enfant.
Quand devriez-vous aller chez le médecin?
Les personnes atteintes du syndrome de Meckel-Gruber présentent déjà de graves problèmes de santé à la naissance. Des malformations et des dysfonctionnements peuvent souvent être détectés pendant le processus de naissance. De nombreux patients atteints sont nés avec une fente labiale et palatine et doivent recevoir des soins médicaux dès que possible. Dans le cas d'une naissance hospitalière, les infirmières et médecins présents prennent en charge les premiers soins du nouveau-né. Souvent, une intervention chirurgicale immédiate est ordonnée pour assurer la survie de l'enfant. Les sages-femmes et les obstétriciens assument ces tâches dans le cas d'un accouchement à domicile ou d'un accouchement dans une maison de naissance. Il est de votre responsabilité d'organiser un transport de l'enfant vers l'hôpital le plus proche.
Les parents n'ont donc pas à être actifs dans ces formes de naissance. En cas d'accouchement spontané sans la présence de personnel infirmier qualifié, un service ambulancier doit être alerté immédiatement. Si la tête est déformée ou déformée, le crâne est ouvert ou les membres irréguliers, un médecin est nécessaire de toute urgence. Le syndrome est caractérisé par la présence de plus de dix doigts ou orteils. En cas de troubles respiratoires, des mesures de premiers soins supplémentaires sont nécessaires jusqu'à l'arrivée du médecin urgentiste afin que le bébé ne meure pas dans les premières minutes de sa vie. La survie ne peut être assurée que par la réanimation bouche-à-bouche.
Thérapie et traitement
Le syndrome de Meckel-Gruber ne peut être traité. Si un diagnostic est posé avant la naissance, une interruption de grossesse est souvent envisagée. En raison des graves malformations du crâne et des organes, le taux de mortalité dans le syndrome de Meckel-Gruber est de 100%, ce qui signifie que tous les nouveau-nés atteints ne sont pas viables à long terme. La plupart des enfants meurent dans les sept premiers jours; aucun enfant ne vit généralement plus de deux semaines.
Perspectives et prévisions
Le pronostic du syndrome de Meckel-Gruber est extrêmement défavorable. Le syndrome est basé sur un défaut génétique. Cela ne peut pas être guéri avec les possibilités médicales actuelles. L'enfant naît avec de graves handicaps et a peu de chances de survie. En raison des exigences légales, les médecins et les chercheurs ne sont en aucun cas autorisés à modifier la génétique humaine. En conséquence, les médecins ne peuvent se concentrer que sur l'utilisation d'options de traitement qui soulagent les divers symptômes.
Cependant, étant donné que plusieurs restrictions sanitaires sévères doivent être documentées pour cette maladie, les options de traitement actuelles ne sont pas suffisantes pour stabiliser la personne touchée. Quelques jours ou semaines après l'accouchement, tous les cas connus de syndrome de Meckel-Gruber entraînent une mort prématurée du patient. Indépendamment de la précocité du diagnostic et de la rapidité avec laquelle des mesures médicales étendues sont prises, l'espérance de vie moyenne de la personne touchée se situe entre une et deux semaines après la naissance.
Les malformations physiques affectent plusieurs zones du système squelettique et des organes. Le corps du nouveau-né est trop faible pour survivre aux nombreuses opérations nécessaires à la stabilisation de l'organisme. Par conséquent, malgré tous les efforts, une défaillance d'organe et donc une mort prématurée se produiront inévitablement.
la prévention
En principe, le syndrome de Meckel-Gruber ne peut être évité. Un diagnostic précoce n'empêche pas la maladie, mais permet seulement une interruption précoce de la grossesse. Les chercheurs ont identifié trois sites génétiques où les changements pourraient être responsables de la maladie héréditaire grave. Ces emplacements sont appelés locus de fièvre aphteuse:
- MKS1 est sur le chromosome 17
- MKS2 sur le chromosome 11
- MKS3 sur le huitième chromosome.
Des changements dans le gène FMD3 ont été détectés chez des personnes originaires du Pakistan. Des modifications du gène de la fièvre aphteuse ont également eu lieu en Finlande et en Europe. Une modification du gène de la fièvre aphteuse a jusqu'à présent été identifiée comme une cause définitive de la maladie. Il existe un diagnostic génétique qui vérifie la présence de ce gène défectueux. La condition préalable à la réalisation de ce diagnostic est qu'un diagnostic fiable du syndrome de Meckel-Gruber ait déjà été posé.
Si le syndrome de Meckel-Gruber peut également être détecté avec un diagnostic génétique chez cet enfant malade, un diagnostic génétique peut être effectué avant la naissance lors de grossesses ultérieures. De cette façon, les parents peuvent savoir avec certitude si leur enfant à naître est porteur du défaut génétique. Il est également possible de tester les parents s'ils soupçonnent la fièvre aphteuse. Un test sanguin est effectué pour déterminer si vous êtes porteur du défaut génétique et s'il existe un risque de transmission de la maladie à la future progéniture.
Suivi
En règle générale, le syndrome de Meckel-Gruber réduit également considérablement l'espérance de vie des enfants, de sorte qu'ils ne meurent que quelques semaines après la naissance. Les soins de suivi se concentrent sur les endeuillés.
Parfois, un soutien psychologique professionnel peut aider à gérer la perte de l'enfant et le chagrin. En collaboration avec les personnes touchées, cela développe des mesures thérapeutiques pour absorber le stress psychologique sévère. Si les parents déjà atteints souhaitent avoir à nouveau des enfants, nous leur recommandons de consulter le gynécologue traitant afin de déterminer à l'avance la probabilité d'un autre enfant malade.
Tu peux le faire toi-même
Le syndrome de Meckel-Gruber est une maladie grave qui, dans la grande majorité des cas, entraîne la mort de l'enfant. En raison de ce pronostic négatif, la mesure d'auto-assistance la plus importante est de rechercher un soutien thérapeutique. Le gynécologue peut orienter les parents vers un spécialiste approprié, avec l'aide duquel les peurs et les inquiétudes typiques peuvent être discutées et traitées. Il est également recommandé d'assister à un groupe d'entraide. Parler à d'autres parents touchés facilite la gestion de la maladie et de son issue principalement négative.
En collaboration avec le gynécologue responsable, il faut également décider si l'enfant doit être porté à terme ou avorté. Habituellement, les parents décident de se faire avorter parce que les chances de guérison sont très minces, mais dans certains cas, une naissance normale est également possible et raisonnable. En alternative, il existe une livraison palliative.
Quel que soit le choix des parents, un soutien psychologique et l'aide d'amis et de parents sont nécessaires. Si la femme souhaite par la suite redevenir enceinte, un examen complet doit être effectué pour déterminer les chances d'avoir un enfant en bonne santé.
.jpg)


.jpg)
.jpg)





.jpg)


.jpg)