le Syndrome de Mowat-Wilson est un trouble génétique rare du développement avec un large éventail de symptômes. En plus des anomalies faciales, intestinales et génitales, des malformations cardiaques et des troubles du développement cérébral surviennent dans le cadre du défaut génétique. La maladie jusqu'ici incurable ne peut être traitée que de manière symptomatique.
Qu'est-ce que le syndrome de Mowat-Wilson?

© Jezper - stock.adobe.com
le Syndrome de Mowat-Wilson est un tableau clinique assez jeune. Le phénomène de diversité clinique a été décrit pour la première fois par Mowat et Wilson en 1998. Outre les troubles du développement, la microcéphalie et le complexe des symptômes de la maladie de Hirschsprung caractérisent le tableau clinique. Un défaut génétique est considéré comme la cause de la maladie.
Dans l'ensemble, les symptômes sont extrêmement divers. Jusqu'à présent, la maladie rare a fait l'objet de peu de recherches. En conséquence, peu d'options thérapeutiques sont disponibles à ce jour. Il n'y a pas de prévalence définitive car le trouble pourrait rarement ou pas du tout être diagnostiqué bien avant le 21e siècle. Il y a actuellement environ 200 patients documentés atteints du syndrome.
causes
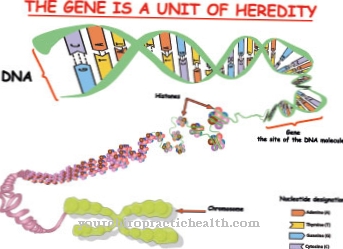
Une mutation génétique provoque le syndrome de Mowat-Wilson. Selon des recherches récentes, le gène ZFHX1B est le gène responsable de la maladie. Le défaut génétique causal serait dans la région chromosomique 2q22. Le gène affecté a une taille d'environ 70 kb et se compose d'un total de dix exons de 1214 acides aminés. Ce gène code pour la protéine SIP1, qui est active en tant que modulateur de transcription et est impliquée dans l'embryogenèse.
L'embryogenèse des personnes atteintes est donc perturbée. Les anomalies pathogènes du gène peuvent correspondre à une délétion complète, un repositionnement ou une anomalie séquentielle. Le défaut génétique est transmis dans l'hérédité autosomique dominante. Un allèle défectueux sur les deux chromosomes homologues suffit à transmettre la maladie héréditaire.
Symptômes, maux et signes
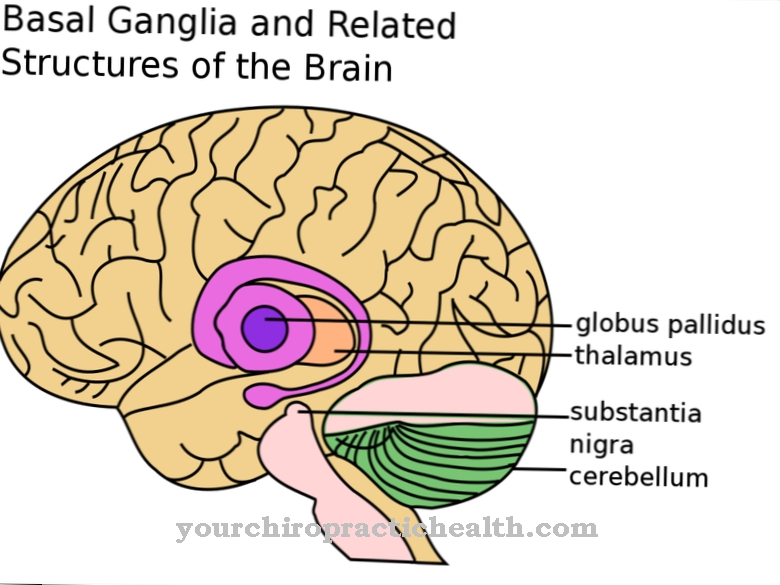
Les symptômes du syndrome de Mowat-Wilson correspondent à un trouble du développement complexe et sont cliniquement diversifiés. Les principaux symptômes comprennent des convulsions déclenchées par le cerveau et une microcéphalie. Une telle microcéphalie se produit à la suite du durcissement prématuré de toutes les sutures crâniennes et resserre le cerveau pendant la phase de croissance. Pour cette raison, les patients souffrent d'un retard mental. De plus, il existe souvent des anomalies du visage qui donnent souvent au patient un profil en forme d'aigle.
Ces anomalies peuvent inclure, par exemple, de grands yeux enfoncés, des sourcils pointés horizontalement, des anomalies de l'oreillette, des lobes d'oreille incarnés et un menton proéminent. Dans 90% des cas, les personnes atteintes souffrent d'épilepsie. Le développement mental est gravement retardé et le développement linguistique est souvent complètement absent. Le développement moteur du patient est également ralenti.
Avec des mesures de naissance normales, une petite taille secondaire se produit souvent. Il peut y avoir des malformations de l'urètre. Des malformations cardiaques congénitales ou des malformations des organes génitaux sont également envisageables. De plus, des anomalies neuronales du plexus de la paroi intestinale se produisent, car elles sont caractéristiques de la maladie de Hirschsprung.
Diagnostic et évolution de la maladie
Le diagnostic du syndrome de Mowat-Wilson ne peut être posé sur la base de simples examens, mais nécessite une analyse du matériel génétique. Le laboratoire amplifie les exons deux à dix du gène ZFHX1B à partir de l'ADN génomique du patient. Cette amplification a lieu par PCR. L'analyse de ce matériel et des sites d'épissage intron-exon a lieu par séquençage d'ADN.
Chaque exon du gène ZFHX1B est examiné pour la délétion et la duplication via une amplification de sonde dépendante de la ligature multiplex. Cette procédure élaborée dure environ trois semaines et, contrairement au simple examen du patient, peut permettre un diagnostic sans équivoque. Dans la plupart des cas, en plus de l'ADN de la personne, celui de ses parents est également séquencé et analysé.
L'évolution de la maladie dépend fortement de la forme de l'anomalie génétique et de l'étendue de la suppression ou du repositionnement des parties chromosomiques. Des pronostics définitifs peuvent difficilement être établis en raison des cas de maladie peu documentés à ce jour. Cependant, un diagnostic précoce et un traitement ultérieur auront probablement une influence positive sur le pronostic.
Complications
Le syndrome de Mowat-Wilson provoque de graves plaintes et complications chez le patient, ce qui réduit considérablement l'espérance de vie et la qualité de vie. En règle générale, la vie quotidienne du patient est également considérablement limitée et les personnes touchées dépendent de l'aide d'autres personnes dans leur vie quotidienne.
En outre, il existe un retard mental dans lequel les proches et les parents souffrent souvent de troubles psychologiques ou de dépression. Dans la plupart des cas, les personnes touchées souffrent également de crampes et d'une diminution de la résilience. En outre, diverses déformations du visage se produisent également et une épilepsie se produit.
Le développement linguistique est également considérablement retardé, de sorte qu'à l'âge adulte, il y a des difficultés considérables à communiquer avec le patient. Une malformation cardiaque et une petite taille se produisent également. La malformation cardiaque peut entraîner une mort cardiaque spontanée, de sorte que l'espérance de vie de la personne touchée est limitée par le syndrome de Mowat-Wilson.
Il n'existe aucun remède pour le syndrome de Mowat-Wilson. Cependant, les différentes plaintes peuvent être limitées et traitées afin que la personne concernée ait une vie quotidienne supportable. Il n'y a pas de complications, mais un traitement positif n'est pas toujours possible.
Quand devriez-vous aller chez le médecin?
Bien que le syndrome de Mowat-Wilson ne puisse pas être guéri avec les options légales et médicales actuelles, le traitement des symptômes qui surviennent peut apporter un soulagement significatif. Normalement, plus le diagnostic peut être posé tôt, meilleures sont les options thérapeutiques du patient. La consultation d'un médecin est nécessaire si un trouble du développement survient chez l'enfant en pleine croissance.
S'il existe des anomalies individuelles en comparaison directe avec celles du même âge, un médecin est nécessaire. Les observations doivent être discutées avec lui afin qu'une évaluation de l'état de santé soit possible. Un médecin doit être présenté avec un trouble d'apprentissage, une déficience de la mémoire, un retard de la parole ou des particularités des séquences de mouvement. Si vous ressentez des crampes, des douleurs ou une posture anormale, vous devriez consulter un médecin. Les malformations ou anomalies faciales indiquent une condition qui nécessite un traitement.
Un défaut visuel ou des anomalies des traits du visage doivent être clarifiés par un médecin. Les processus de pensée ou les mouvements ralentis sont des signes d'un trouble et doivent être étudiés. En cas de troubles du rythme cardiaque, de problèmes d'excrétions ou d'irrégularités dans la capacité à réagir ou à percevoir, un médecin doit être consulté. Les troubles du comportement, les troubles végétatifs ou les particularités de l'apparence de la peau doivent être examinés par un médecin.
Traitement et thérapie
Le syndrome de Mowat-Wilson est jusqu'à présent incurable. Les options de traitement symptomatique sont également limitées. Les thérapies médicamenteuses sont généralement utilisées contre les crises. Les antiépileptiques montrent la plus grande efficacité dans ce contexte. Certaines des malformations symptomatiques peuvent être corrigées chirurgicalement. En particulier, les symptômes de la maladie de Hirschsprung doivent être corrigés le plus tôt possible, faute de quoi une septicémie ou une péritonite pourraient s'installer.
Le traitement symptomatique du syndrome de Mowat-Wilson vise principalement à améliorer la qualité de vie des personnes touchées. Dans ce but, le retard mental et moteur peut également être contrecarré. Les thérapies orthophoniques peuvent dans certaines circonstances contribuer au développement du langage, qui dans le syndrome de Mowat-Wilson échoue souvent complètement sans mesures thérapeutiques de soutien. Les traitements physiothérapeutiques et ergothérapiques peuvent contrer le retard de développement des habiletés motrices.
Le syndrome de Mowat-Wilson est souvent un fardeau psychologique presque inimaginable pour les parents d'une personne touchée. Pour cette raison, les parents des patients sont souvent pris en charge par des psychothérapeutes. La recherche médicale s'intéresse actuellement aux approches de thérapie génique qui devraient guérir les défauts génétiques à l'avenir. De cette manière, le gène défectueux ZFHX1B chez les personnes touchées peut bientôt être remplacé, ce qui peut rendre la maladie curable.
Perspectives et prévisions
Le syndrome de Mowat-Wilson peut être bien traité de nos jours. L'espérance de vie et la qualité de vie sont basées sur le type et la gravité des malformations congénitales. Avec des anomalies légères qui n'affectent pas le cœur, les personnes touchées peuvent vivre jusqu'à l'âge adulte.
Les patients gravement malades meurent généralement pendant l'enfance ou l'adolescence des suites de la maladie. Les causes typiques de décès sont l'infarctus du myocarde ou les maladies HSCR caractéristiques. Les crises cérébrales entraînent souvent la mort au cours des premières années de vie de l'enfant. Le syndrome rare peut être traité de manière symptomatique, ce qui signifie que les patients peuvent mener une vie sans symptômes, au moins temporairement.
À long terme, cependant, le syndrome de Mowat-Wilson n'offre pas de pronostic positif, car les différentes malformations et anomalies entraînent une détérioration progressive de la santé et aboutissent finalement à la mort. Le pronostic de l'espérance de vie et de l'évolution de la maladie est généralement établi par le spécialiste responsable. La plupart du temps, il s'agit d'un neurologue ou d'un spécialiste des maladies génétiques. Selon les symptômes, le diagnostic de la maladie peut être difficile, c'est pourquoi le syndrome de Mowat-Wilson n'est souvent pas diagnostiqué avant que la maladie ne soit bien avancée.
la prévention
Comme le syndrome de Mowat-Wilson est un trouble du développement complexe avec une cause génétique, le phénomène peut difficilement être évité. Les couples impliqués dans la planification familiale peuvent cependant faire séquencer leur ADN afin d'évaluer leur risque personnel de transmettre des anomalies génétiques.
Suivi
Dans la plupart des cas, les personnes atteintes du syndrome de Mowat-Wilson ont peu ou pas de soins de suivi disponibles, car il s'agit d'une maladie génétique. Par conséquent, les personnes concernées devraient idéalement consulter un médecin à un stade précoce afin qu'il n'y ait plus de plaintes ou de complications qui pourraient avoir un impact négatif sur l'espérance de vie et la qualité de vie de la personne concernée.
En règle générale, l'auto-guérison ne peut pas se produire, de sorte qu'un médecin doit être consulté dès les premiers signes et symptômes de la maladie. Si vous souhaitez avoir des enfants, des tests génétiques et des conseils peuvent être utiles pour éviter que le syndrome ne se reproduise chez vos descendants. En règle générale, les personnes atteintes du syndrome de Mowat-Wilson dépendent de la prise de divers médicaments.
Ceux-ci doivent toujours être pris à temps et à la dose correcte pour soulager les symptômes. Dans le cas des enfants, les parents en particulier devraient contrôler l'apport. Des mesures de physiothérapie sont également nécessaires dans de nombreux cas, bien que certains des exercices puissent également être pratiqués chez vous. On ne peut pas prédire universellement si le syndrome de Mowat-Wilson entraînera une réduction de l'espérance de vie de la personne touchée.
Tu peux le faire toi-même
Puisqu'il n'existe malheureusement pas de remède pour le syndrome de Mowat-Wilson, la principale priorité est actuellement d'améliorer la qualité de vie de l'enfant.
Dans de nombreux cas, l'orthophonie commencée tôt peut contrecarrer le retard du développement linguistique et assurer un succès significatif dans le développement du langage. De plus, des mesures intensives de physiothérapie et d'ergothérapie assurent un meilleur développement moteur et mental. En plus des mesures prescrites par le médecin, il est également conseillé de traiter le sujet vous-même et de poursuivre la thérapie à la maison.
Prendre soin d'un enfant handicapé est un fardeau énorme, en particulier pour les parents mais aussi pour les frères et sœurs qui peuvent être présents, ce qui peut affecter la vie de famille et finalement la qualité des soins. Il est donc extrêmement important que dans de tels cas les parents recherchent une psychothérapie à temps qui leur donne plus de force sur le long terme en apprenant des méthodes de relaxation et de gestion des conflits.
Il convient également de garder à l'esprit que les personnes concernées ont droit à des soins préventifs d'une durée maximale de six semaines par an, dont l'assurance soins prend en charge les frais. Il existe déjà des installations qui fournissent des soins intensifs pendant la journée tandis que les proches peuvent se détendre lors des excursions. Cela peut être d'une grande aide, en particulier avec les frères et sœurs.


.jpg)
.jpg)

.jpg)
.jpg)





.jpg)


.jpg)