Sous un Phéochromocytome une tumeur du marché surrénalien est comprise. Il est capable de produire des hormones.
Qu'est-ce qu'un phéochromocytome?

© SciePro - stock.adobe.com
À Phéochromocytome c'est une tumeur de la médullosurrénale. Dans la plupart des cas, la tumeur productrice d'hormones est bénigne. Les hormones produites sont principalement l'adrénaline et la noradrénaline. Dans 85% des cas, la tumeur se trouve sur la glande surrénale. Alors que 85 pour cent de tous les phéochromocytomes sont de nature bénigne, 15 pour cent développent une évolution maligne. 90% de la tumeur est unilatérale. Avec les 10% restants, il s'installe des deux côtés.
Le paragangliome est une forme rare de phéochromocytome. La tumeur longe le tronc sympathique à l'extérieur des glandes surrénales. Ceci est situé dans la région de l'estomac et de la poitrine dans la région de la colonne vertébrale. Le phéochromocytome est rare. Deux à huit cas de maladie surviennent chez environ un million de personnes. Les adultes sont particulièrement touchés par la maladie, mais les enfants souffrent aussi parfois de la tumeur.
causes
La cause du développement d'un phéochromocytome ne peut pas toujours être découverte. En médecine, on parle d'un phéochromocytome sporadique. Ce type de tumeur apparaît principalement entre 40 et 50 ans. Certaines formes de phéochromocytome sont héréditaires. Ils résultent de mutations au sein du génome et surviennent principalement chez les personnes plus jeunes de moins de 40 ans. Environ 10 pour cent de ces phéochromocytomes apparaissent dans les familles.
Dans certains cas, le phéochromocytome est associé à d'autres maladies héréditaires. Ceux-ci incluent le syndrome de von Hippel Lindau, la néoplasie endocrinienne multiple de type 2 et la neurofibromatose de type 1. Une caractéristique typique du phéochromocytome est la production accrue d'hormones de stress telles que l'adrénaline et la noradrénaline. La tumeur peut également produire de la dopamine, mais c'est moins fréquent.
Symptômes, maux et signes
La libération accrue d'adrénaline et de noradrénaline entraîne les symptômes du phéochromocytome. Cependant, les plaintes varient d'une personne à l'autre. Certains patients ne présentent même aucun symptôme, ce qui signifie que la tumeur n'est découverte que par hasard. L'hypertension artérielle est un symptôme typique du phéochromocytome. Cela conduit à une augmentation brutale de la pression artérielle, qui peut même mettre la vie en danger.
Les médecins parlent alors de crise hypertensive ou de crise de tension artérielle. Environ 50 pour cent de tous les patients sont affectés par ces symptômes. Parfois, la pression artérielle peut également être chroniquement élevée, ce qui est connu sous le nom d'hypertension persistante. Environ la moitié de tous les patients adultes et environ 90% de tous les enfants en souffrent.
Mais il existe d'autres symptômes de phéochromocytome possibles. Ceux-ci incluent la perte de poids, l'angine de poitrine, les maux de tête, les sueurs, les battements cardiaques, les tremblements, l'anxiété et la pâleur du visage. Le niveau élevé d'adrénaline entraîne également une augmentation de la glycémie. Il existe un risque accru de développer un diabète sucré à mesure que la maladie progresse.
Diagnostic et évolution de la maladie
Le diagnostic d'un phéochromocytome est généralement basé sur les symptômes typiques. Les crises d'hypertension occasionnelles, où les traitements médicamenteux habituels sont inefficaces, sont principalement considérées comme suspectes. Il est également important de mesurer les hormones du stress, l'adrénaline et la noradrénaline. Pour une mesure fiable, soit un échantillon d'urine, qui est prélevé 24 heures, soit un examen du plasma sanguin dans les 30 minutes.
En cas d'augmentation de la teneur en hormones, un test d'inhibition de la clonidine est effectué. La clonidine est l'une des substances actives contre l'hypertension artérielle. Alors que la clonidine inhibe la libération d'adrénaline chez les personnes en bonne santé, la valeur reste élevée dans le cas d'un phéochromocytome. Des méthodes d'imagerie telles que la tomodensitométrie (TDM) ou l'imagerie par résonance magnétique (IRM) sont utilisées pour détecter un phéochromocytome.
Une scintigraphie MIBG spéciale est réalisée pour localiser une tumeur située à l'extérieur de la glande surrénale. L'évolution de la maladie dans un phéochromocytome est différente. Chez environ 80% de tous les patients, la pression artérielle revient à la normale après que la tumeur a été enlevée avec succès. Cependant, le reste des patients souffrent d'hypertension car d'autres causes jouent un rôle. Le phéochromocytome se reproduit après une longue période chez environ 15 pour cent de toutes les personnes touchées.
Complications
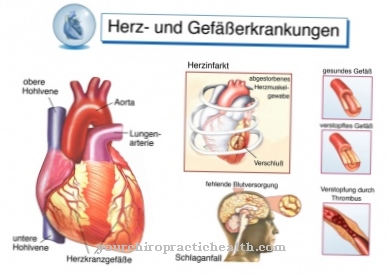
Les symptômes du phéochromocytome peuvent être très différents. Pour cette raison, les complications et les symptômes ne peuvent généralement pas être prédits universellement. Dans la plupart des cas, cependant, cette maladie entraîne une hypertension artérielle. Les personnes touchées peuvent également souffrir d'une crise cardiaque, qui, dans le pire des cas, entraîne la mort.
Un malaise général et une hypertension peuvent également survenir et ont généralement un effet très négatif sur la vie quotidienne du patient. Il n'est pas rare que les personnes touchées souffrent d'anxiété ou de transpiration. La maladie peut également provoquer un rythme cardiaque rapide ou des maux de tête sévères. De plus, il y a aussi une pâleur du visage et des palpitations. Si le phéochromocytome n'est pas traité, il peut également conduire au diabète.
Le traitement du phéochromocytome consiste généralement à retirer la tumeur à l'origine des symptômes. On ne peut pas prédire universellement si cela entraînera des complications.La chimiothérapie est également généralement nécessaire. Le phéochromocytome peut également réduire l'espérance de vie de la personne.
Quand devriez-vous aller chez le médecin?
Comme un phéochromocytome est une tumeur, il doit être traité immédiatement par un médecin. Plus le traitement et le diagnostic sont précoces, plus les chances de guérison complète sont élevées. Souvent, la tumeur elle-même n'est découverte que par hasard, car elle ne conduit à aucun symptôme clair. Après le diagnostic, cependant, il doit être retiré rapidement. Dans de nombreux cas, le phéochromocytome se manifeste par une hypertension artérielle. Si l'hypertension artérielle survient sans raison particulière et, surtout, de façon permanente, un médecin doit être consulté. La pression artérielle elle-même est souvent élevée de façon chronique. Le phéochromocytome est également perceptible par une perte de poids, une pâleur du visage ou par de fortes sueurs. Consultez un médecin si vous présentez l'un de ces symptômes.
En règle générale, le premier examen peut être effectué par le médecin de famille. Cependant, un examen plus détaillé et éventuellement un traitement nécessiteront un médecin spécialiste. On ne peut généralement pas prédire si le phéochromocytome conduira à une espérance de vie réduite pour le patient.
Thérapie et traitement
L'ablation chirurgicale de la tumeur est considérée comme le meilleur traitement du phéochromocytome. La plupart des patients subissent une laparoscopie mini-invasive à cet effet. Seules trois incisions plus petites doivent être pratiquées sur la paroi abdominale. Après avoir inséré les instruments chirurgicaux, le chirurgien retire enfin la tumeur. Si la tumeur est plus grosse ou difficile à enlever, une laparotomie plus étendue peut être nécessaire.
Si les deux glandes surrénales sont affectées par un phéochromocytome, cela se traduit par un déficit en hormone stéroïde après une opération. Pour compenser cela, les hormones manquantes sont remplacées par des médicaments. Le traitement ultérieur dépend du fait que le phéochromocytome est bénin ou malin. Une tumeur maligne peut entraîner le développement de métastases (tumeurs filles). La chimiothérapie ou la radiothérapie à l'iode sont envisageables comme mesures thérapeutiques possibles.
Parfois, il n'est pas possible de traiter chirurgicalement un phéochromocytome. Dans de tels cas, une thérapie symptomatique a lieu. Cela traite les plaintes qui surviennent en raison de la production excessive d'hormones de stress. Par exemple, les alpha-bloquants peuvent réduire l'effet d'adrénaline en bloquant les points d'ancrage.
Perspectives et prévisions
Le pronostic d'un phéochromocytome varie et dépend de plusieurs facteurs. S'il est possible d'opérer la tumeur à un stade précoce et si le patient ne souffre pas de maladies supplémentaires, les perspectives sont généralement favorables. Les symptômes disparaissent souvent à nouveau. Chez environ 50 à 80 pour cent de toutes les personnes touchées, la pression artérielle revient à la normale après une intervention chirurgicale, à condition qu'il s'agisse d'un phéochromocytome bénin.
Si le patient souffre de phéochromocytome depuis longtemps, il existe un risque de symptômes secondaires tels qu'une insuffisance cardiaque en raison de l'augmentation de la pression artérielle. Environ 15 pour cent de toutes les personnes touchées doivent s'attendre à ce qu'un phéochromocytome se reproduise après le traitement. Les médecins parlent alors d'une rechute. Pour cette raison, le patient doit subir des contrôles à intervalles réguliers. Ils servent à détecter une éventuelle rechute à temps pour qu'un traitement efficace puisse être effectué.
Si un phéochromocytome bénin est enlevé chirurgicalement, une hypertension essentielle existe toujours. Cela signifie que la pression artérielle continuera d'être élevée car d'autres déclencheurs sont également présents. Si un phéochromocytome bénin est présent, 95% de tous les patients survivront aux cinq prochaines années. Si, cependant, il s'agit d'une tumeur maligne et que des métastases se sont déjà formées, le taux de survie à 5 ans tombe à environ 44%.
la prévention
La prévention d'un phéochromocytome n'est pas possible. Cependant, le conseil génétique peut être utile pour les familles avec une prédisposition.
Suivi
Avec un phéochromocytome, la personne affectée n'a que des mesures et des options de suivi très limitées dans la plupart des cas. Puisqu'il s'agit d'une tumeur aux reins, un médecin doit être contacté très tôt afin qu'il n'y ait pas de complications ou d'autres plaintes dans le cours ultérieur. Plus tôt un médecin est consulté, meilleure est l'évolution de la maladie, en règle générale, car aucune guérison spontanée ne peut se produire.
La plupart des personnes touchées dépendent d'une opération. La personne touchée doit absolument se reposer après une telle opération et prendre soin de son corps. L'effort ou les activités stressantes et physiques doivent être évités afin de ne pas continuer à alourdir le corps. La plupart des personnes atteintes de phéochromocytome dépendent également du soutien des amis et de la famille dans la vie quotidienne.
Il n'est pas rare que le soutien psychologique soit également très important pour éviter tout bouleversement psychologique ou dépression. Des contrôles et des examens réguliers par un médecin sont également très importants afin d'identifier et d'éliminer d'autres tumeurs à un stade précoce. Dans certains cas, le phéochromocytome réduit l'espérance de vie des personnes touchées.
Tu peux le faire toi-même
Un phéochomocytome est généralement enlevé chirurgicalement. Les mesures que le patient peut prendre par la suite pour récupérer rapidement dépendent de l'emplacement de la tumeur et d'autres facteurs.
En principe, rien ne doit être mangé dans les premières heures suivant l'intervention. Après une anesthésie générale, vous devez également vous abstenir de boire. Après un jour, le patient est autorisé à manger à nouveau des aliments fades. Le médecin établira des directives précises sur la nutrition et, si nécessaire, impliquera un nutritionniste. Pour compenser la perte de nutriments et de liquides, vous devez manger et boire suffisamment dans les jours et les semaines suivant l'opération du phéochomocytome. Des compléments alimentaires et des solutions d'infusion sont également disponibles. L'alcool doit être évité au cours des premières semaines car il a un effet négatif sur la coagulation sanguine.
De plus, la protection physique s'applique toujours. La partie du corps qui a été opérée est particulièrement sensible à la douleur et ne doit pas être exposée à de fortes vibrations. Au lieu de cela, un refroidissement soigneux est recommandé. Une fois le phéochomocytome retiré, des symptômes supplémentaires peuvent apparaître initialement. Celles-ci devraient s'être calmées au plus tard au bout de quelques semaines. Si les symptômes persistent, il est préférable d'en informer le médecin.
.jpg)




.jpg)

.jpg)







.jpg)