le Syndrome de Maroteaux-Lamy appartient aux mucopolysaccharidoses, qui comprennent diverses maladies du stockage lysosomal. Le syndrome est dû à une mutation génétique qui entraîne une activité enzymatique insuffisante et conduit à des dépôts de dermatosulfates. La thérapie consiste principalement en des thérapies de remplacement enzymatique.
Qu'est-ce que le syndrome de Maroteaux-Lamy?

© imaginuma - stock.adobe.com
Les mucopolysaccharidoses sont un groupe indépendant de maladies qui contiennent des maladies de stockage lysosomal. Les maladies de stockage lysosomal existent au coin de la rue. Toutes sont des maladies métaboliques génétiquement déterminées qui sont causées par un dysfonctionnement du lysosome. L'une de ces maladies est la soi-disant Syndrome de Maroteaux-Lamy. Le trouble métabolique inné conduit à un stockage de dermatinsulfates.
Les termes sont utilisés comme synonymes Mucopolysaccharidose de type VI, Déficit en aryl sulfatase B, Déficit ARSB et Déficit en ASB tel que Déficit en N-acétylgalactosamine-4-sulfatase. La maladie a été décrite pour la première fois en 1963. Les généticiens et pédiatres humains parisiens P. Maroteaux et M. Lamy sont considérés comme les premiers à la décrire.
La prévalence de la maladie se situe entre une et neuf personnes touchées sur 100 000 personnes. Une accumulation familiale a été observée dans les cas documentés jusqu'à présent. Le syndrome est hérité de manière autosomique récessive. Une mutation génétique est considérée comme la cause de la maladie.
causes
Le syndrome de Maroteaux-Lamy est dû à une mutation essentiellement héréditaire. Les mutations causales ont maintenant été localisées sur le gène ARSB. On dit que des mutations au locus du gène 5q13 à 5q14.1 peuvent provoquer le complexe de symptômes. Les gènes qui s'y trouvent codent dans l'ADN pour une protéine spécifique.
On dit qu'une mutation des gènes entraîne une activité anormale de l'arylsulfatase B mutée, également connue sous le nom d'ASB ou N-acétylgalactosamine-4-sulfatase. L'activité de la substance est réduite dans le cadre de la mutation. En raison de cette réduction, il y a des perturbations dans la décomposition de substances telles que le sulfate de chondroïtine et le sulfate de dermatane. Comme les substances ne sont plus suffisamment décomposées en raison de la mutation causale, le corps stocke les résidus des substances.
Les symptômes typiques du syndrome de Maroteaux-Lamy résultent de ce stockage. On ne sait pas encore si les facteurs génétiques ainsi que les influences externes jouent un rôle dans le développement de la mutation. Cela peut être supposé, au moins pour les nouvelles mutations.
Symptômes, maux et signes
Les patients atteints du syndrome de Maroteaux-Lamy souffrent d'un ensemble de critères cliniquement caractéristiques. L'un des symptômes les plus importants est une petite taille agissant de manière disproportionnée, caractérisée par un tronc court. La disproportion du patient est associée à un visage grossier qui rappelle les symptômes de la maladie de Hurler.
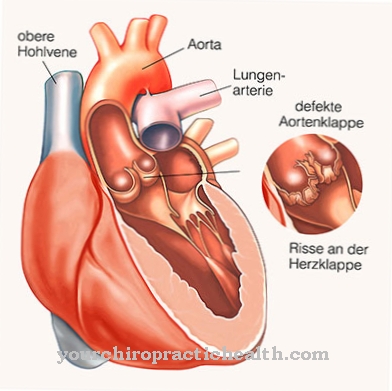
Dans la plupart des cas, la cornée du patient est trouble. De plus, il peut y avoir des hépatosplénomégalies, des hernies ou des contractures des articulations. Les valvules cardiaques des patients s'épaississent de plus en plus en raison des dépôts. En outre, il peut y avoir une dysplasie squelettique similaire à celle des patients atteints de dysostose multiplex.
Le tableau clinique et l'évolution du syndrome sont considérés comme variables et individuels. En plus des processus lents, des processus rapides ont également été documentés. Si les premiers symptômes se manifestent immédiatement après la naissance, ce phénomène parle d'une évolution assez rapide.
Cela est particulièrement vrai pour une augmentation du glycosaminoglycane dans l'urine, pour une dysostose multiplex sévère et une petite taille manifeste. Les personnes touchées dont l'évolution est lente présentent généralement les premiers symptômes beaucoup plus tard. L'augmentation de GAG est beaucoup plus faible et le multiplex de dysostose est beaucoup plus doux.
Diagnostic et évolution de la maladie
Le syndrome de Maroteaux-Lamy ne doit pas nécessairement se manifester immédiatement après la naissance et n'est dans de nombreux cas diagnostiqué que plus tard, lorsque les premiers symptômes apparaissent. Le diagnostic est basé sur les critères cliniquement typiques de la maladie et repose donc principalement sur une activité ASB significativement réduite, qui peut être tracée sur des fibroblastes et des leucocytes cultivés.
En revanche, une activité normale existe par rapport aux autres sulfatases. Dans le cadre du diagnostic, le médecin fournit également des preuves de l'augmentation de la quantité de sulfate de dermatane excrétée dans l'urine. Dans le diagnostic différentiel, le syndrome de Maroteaux-Lamy doit être différencié du déficit multiple en sulfatases et des autres formes de mucopolysaccharidose. La sialidose et la mucolipidose sont également des maladies importantes pour le diagnostic différentiel. Le pronostic du patient varie d'un cas à l'autre et dépend principalement de l'âge d'apparition et de la gravité des premiers symptômes.
Complications
Tout d'abord, le syndrome de Maroteaux-Lamy conduit à une petite taille du patient. Les enfants en particulier peuvent souffrir d'intimidation et de taquineries à un jeune âge et développer des problèmes psychologiques ou une dépression en conséquence. En règle générale, la croissance du patient ne se fait pas proportionnellement et diverses plaintes et malformations se produisent, qui se produisent également au visage.
De plus, les valves cardiaques sont endommagées et déplacées par le syndrome de Maroteaux-Lamy, de sorte qu'il y a des plaintes ou des limitations dans le cœur. Dans certains cas, cela peut également réduire l'espérance de vie du patient, ce qui peut entraîner une mort cardiaque. Un traitement causal de ce syndrome n'est pas possible. Pour cette raison, l'objectif principal du traitement est de limiter et de combattre les symptômes afin que la personne touchée puisse mener une vie ordinaire.
Un traitement psychologique peut également être nécessaire. Il n'y a pas de complications particulières, bien que toutes les plaintes ne puissent pas être complètement limitées. En règle générale, l'espérance de vie n'est pas non plus réduite par le syndrome de Maroteaux-Lamy s'il n'y a pas de symptômes cardiaques.
Quand devriez-vous aller chez le médecin?
Si un enfant en pleine croissance montre des signes de perturbations ou de changements dans le processus de développement physique, un médecin doit être consulté. Si l'enfant est clairement de petite taille ou a une malformation du système squelettique, il a besoin d'une aide médicale. S'il y a des anomalies ou des particularités de la forme du corps en comparaison directe avec des enfants du même âge, un médecin doit être consulté pour clarifier les symptômes. Les troubles des séquences de mouvement ou de la motricité générale doivent être présentés à un médecin. Un médecin sera également nécessaire si la cornée est trouble ou si la vue est réduite. Des rythmes cardiaques irréguliers indiquent un problème de santé, qui doit être examiné dans divers tests médicaux.
Dans la plupart des cas, les premières anomalies de la maladie peuvent être reconnues immédiatement après la naissance en raison de modifications du système squelettique. Puisque les nourrissons sont examinés de manière approfondie par le médecin présent après leur naissance, il n'est pas nécessaire que les parents agissent. Si des symptômes tels que des vomissements ou des douleurs apparaissent au cours des premières semaines ou des premiers mois de la vie, un médecin doit être consulté. Si l'enfant pleure et pleure continuellement, c'est le signe d'une irrégularité qui doit être clarifiée. En cas d'essoufflement ou de problème de santé aigu, un service d'urgence doit être alerté. Jusqu'à son arrivée, les premiers soins doivent être fournis pour assurer la survie de l'enfant.
Traitement et thérapie
Une thérapie causale n'est pas disponible pour les patients atteints du syndrome de Maroteaux-Lamy au sens étroit, car la modification de l'activité enzymatique des personnes atteintes est due à une mutation génétique à laquelle seule la thérapie génique pourrait remédier. Cependant, avec la thérapie de remplacement enzymatique au sens le plus large, il existe un type de thérapie qui traite les symptômes de la maladie à leur source.
Dans le traitement enzymatique substitutif, les patients reçoivent de la galsulfase au sens de naglazymes. Ce remplacement enzymatique entraîne une meilleure décomposition des substances concernées et retarde ainsi l'évolution de la maladie. Cependant, les symptômes qui se sont produits jusqu'à présent ne peuvent pas être complètement inversés. L'épaississement des valvules cardiaques peut être traité de manière symptomatique et, dans certaines circonstances, nécessiter un remplacement chirurgical des valvules cardiaques.
Les hernies aiguës sont traitées avec des taxis. L'objectif est une réduction qui laisse au médecin le temps de trouver une solution opératoire. Les opacités cornéennes sévères entraînant la cécité peuvent éventuellement être inversées par une greffe de cornée. Les symptômes tels que la petite taille ne peuvent finalement pas être inversés, mais n'apparaissent souvent que sous une forme légère avec un traitement précoce avec un médicament de remplacement enzymatique.
Vous pouvez trouver votre médicament ici
➔ Médicaments contre la douleurPerspectives et prévisions
Cette maladie rare survient sous un large éventail de formes et de formes.Grâce à l'évolution individuelle et aux différents degrés de syndrome de Maroteaux-Lamy ou de mucopolysaccharidose de type 6, il est généralement difficile de donner un pronostic fiable pour un patient spécifique.
En général, le pronostic est pire si les premiers symptômes du syndrome de Maroteaux-Lamy apparaissent peu après la naissance. Cela signifie généralement une évolution plus rapide de la maladie.
En conséquence, on peut dire que l'âge des personnes atteintes permet de conclure sur le pronostic attendu, tout comme le moment où les premiers symptômes du syndrome de Maroteaux-Lamy sont apparus. En outre, les perspectives du patient dépendent également de la qualité du traitement. Le moment auquel la thérapie de remplacement enzymatique a été initiée est souvent critique.
La thérapie de remplacement enzymatique peut décomposer des substances comme le sulfate de chondroïtine et le sulfate de dermatane. Il ralentit ainsi l'évolution de la maladie. Le problème est que les dommages déjà survenus dans l'organisme ne sont généralement pas réversibles. Cela réduit la qualité de vie, mais peut également entraîner la mort de la personne affectée plus tôt. L'évolution du syndrome de Maroteaux-Lamy peut être rapide si elle survient tôt. Le patient peut également bien répondre au médicament administré. Dans ce cas, le syndrome de Maroteaux-Lamy progressera en conséquence lentement.
la prévention
Jusqu'à présent, aucun facteur externe n'est connu pour le développement du syndrome de Maroteaux-Lamy. La seule mesure préventive actuellement est le conseil génétique en planification familiale.
Suivi
Le traitement du syndrome de Maroteaux-Lamy étant complexe et de longue durée, un suivi classique n'est pas nécessaire. Au contraire, les personnes touchées devraient se concentrer sur la gestion de la maladie en toute sécurité et sur la construction d'une attitude positive malgré l'adversité. Les exercices de relaxation et la méditation peuvent également aider à calmer et à concentrer l'esprit. Étant donné que la petite taille va de pair avec une réduction de l'esthétique, tout complexe d'infériorité et une faible estime de soi doivent être discutés avec un thérapeute si nécessaire. Cela peut aider à mieux accepter la maladie et à améliorer la qualité de vie à long terme.
Tu peux le faire toi-même
Les options d'auto-assistance ne sont pas disponibles pour les personnes atteintes du syndrome de Maroteaux-Lamy. La maladie ne peut être traitée que de manière symptomatique; il n'y a pas de traitement causal.
Afin d'atténuer les symptômes de la maladie, les personnes touchées dépendent de l'apport d'enzymes et de divers médicaments. Ici, un apport régulier et prescrit doit être assuré. Dans les cas graves, cependant, des interventions chirurgicales sur le cœur sont nécessaires. Afin de ne pas stresser inutilement le cœur, les efforts inutiles doivent être évités. Cela est particulièrement vrai pour les charges soudaines ou abruptes.
Si le patient ou les parents veulent à nouveau avoir des enfants, un conseil génétique peut être utile pour éviter que le syndrome de Maroteaux-Lamy ne se reproduise. Les conversations avec des personnes proches ou des amis peuvent souvent soulager les plaintes psychologiques ou la dépression. Le contact avec d'autres patients atteints du syndrome de Maroteaux-Lamy a souvent un très bon effet sur la maladie et peut contribuer à un échange d'informations pouvant éventuellement améliorer la qualité de vie de la personne touchée. Cependant, une guérison complète du syndrome ne peut être obtenue.
.jpg)

.jpg)






.jpg)

.jpg)